Bienvenue dans la 2e partie de Calcul scientifique cloud à l'échelle mondiale avec Kubernetes et Terraform. Je partage ici une démonstration complète du calcul scientifique cloud à l'échelle mondiale avec Kubernetes et Terraform. Pour vous rafraîchir la mémoire ou approfondir les principes et concepts modernes du DevOps, rendez-vous sur la 1re partie de cette série.
Ce guide vous montre comment exploiter concrètement DevOps, conteneurs, Kubernetes et Terraform pour déployer des workloads de calcul.
PerfectScale™ for Kubernetes
Ready to optimize?
Get your free Kubernetes savings analysis
Vue d'ensemble
Dans les grandes lignes, l'exemple de code :
- Montre comment empaqueter des outils bioinformatiques courants (FastQC et BWA) dans des images de conteneurs et les pousser vers le registre d'images de chaque cloud
- Provisionne (et permet de démanteler rapidement) toute l'infrastructure cloud nécessaire à l'exécution des workloads via Terraform, un outil d'Infrastructure as Code
- Déploie un pipeline de workflow standard, basé sur ces images et l'infrastructure cloud provisionnée, vers le service Kubernetes entièrement managé de chaque cloud, un système de gestion et d'orchestration de l'exécution de conteneurs. Argo servira à orchestrer un workflow ou un pipeline de tâches sur un cluster Kubernetes
En combinant Docker, Terraform, Kubernetes et Argo, vous apprendrez à :
- Provisionner, mettre à jour et démanteler l'infrastructure nécessaire à vos workloads avec quelques commandes simples
- Exécuter des workloads analytiques de bout en bout pilotés par DAG, avec relance automatique des étapes individuelles en cas d'erreurs imprévues ou de défaillances d'infrastructure
- Centraliser la journalisation et le suivi des métriques des workloads dans les outils cloud-native d'exploration de logs et de métriques
- Exécuter des workloads sur une infrastructure qui s'adapte automatiquement, à la hausse (jusqu'à une capacité mondiale) comme à la baisse (peu ou aucune ressource de calcul) selon les besoins, afin de ne payer que les ressources CPU/RAM/stockage réellement nécessaires à vos tâches. Fini les serveurs qui tournent à vide et gonflent inutilement la facture
- Déployer en toute fluidité de nouvelles versions logicielles et retirer progressivement les anciennes
Your cloud bill shouldn't be a mystery
Optimization, automation, expertise. In one platform.
Décryptage du code
Maintenant que les bases du DevOps sont posées, voyons comment la base de code associée à cet article illustre les principes fondamentaux du DevOps. Cette démonstration montre comment construire l'infrastructure permettant d'exécuter des pipelines scientifiques à l'échelle mondiale. Concrètement, nous allons exécuter les composants d'un pipeline bioinformatique courant.
Notre objectif :
- Produire des rapports sur une paire de fichiers bruts de séquençage du génome afin de valider leur qualité. Les données brutes de séquençage sont stockées au format FASTQ et les contrôles qualité sont effectués avec un outil nommé FastQC.
- Une fois ces rapports produits, lancer le processus dit d'alignement, qui permettra in fine (via un traitement supplémentaire en aval) de découvrir des mutations. C'est le rôle de l'outil bwa-mem2.
Autrement dit, nous voulons que notre pipeline Kubernetes exécute simultanément deux conteneurs FastQC sur une paire de fichiers de séquençage FASTQ, puis, si ces deux tâches réussissent, lance l'alignement bwa-mem2 de la paire de jeux de données bruts contre la dernière version du génome humain, hg38. Les rapports FastQC ainsi que le fichier d'alignement produit par BWA, appelé fichier BAM, seront déposés dans un bucket pour stockage à long terme.
Attention au coût
Cette base de code vous guide à travers un véritable cas d'usage analytique, et non un simple exemple : la mise en place de l'environnement de calcul aura donc un coût non négligeable. Selon la durée pendant laquelle vous laisserez l'infrastructure Terraform en marche, comptez généralement plusieurs dizaines de dollars sur votre facture cloud.
Étape 0 : Installer les clients locaux : Cloud CLI, Terraform et kubectl
Vérifiez que votre outil CLI cloud est installé localement et authentifié auprès du compte que vous allez utiliser. Liens vers les CLI : GCP et AWS. Provisionner de l'infrastructure-as-code dans le cloud se résume à se placer dans un dossier de fichiers Terraform YAML et à exécuter :
terraform apply -auto-approve
Pour cela, vous devrez installer le client Terraform en local. Terraform réutilisera automatiquement les identifiants de votre CLI cloud.
Lancer un job ou un workflow sur Kubernetes est tout aussi simple : il suffit de créer le template YAML correspondant et d'exécuter :
kubectl create -f job.yaml
Pour cela, vous devrez installer le client kubectl en local. kubectl s'authentifiera via une commande CLI propre à chaque cloud.
Étape 1 : Récupérer les données FASTQ et la référence humaine (hg38)
Avant de démontrer ce workflow, il vous faut une paire de fichiers FASTQ d'exemple ainsi que le génome de référence humain hg38.
Pour limiter les coûts de traitement tout en travaillant sur des volumes proches d'un cas d'usage réel, j'ai récupéré deux fichiers FASTQ d'environ 0,5 Go correspondant à l'échantillon de référence NA12878 depuis le bucket S3 du projet 1000 Genomes. Je recommande vivement de créer un bucket GCS ou S3 dans votre compte cloud et d'y copier ces fichiers : lors de tests répétés du workflow, mieux vaut éviter de retéléchargér les données depuis le bucket S3 1000G à chaque fois.
Gardez toujours un œil sur les tarifs de transfert de données inter-régions .
Les fichiers FASTQ se trouvent dans la région us-east-1 d'Amazon : leur déplacement vers un autre bucket S3 régional (par exemple us-west-2, utilisée pour mes tests) ou hors d'AWS vers un bucket Cloud Storage GCP entraînera des frais pour chaque Go transféré hors de la région du bucket. Créez un bucket dans la région cloud de votre choix et copiez-y les FASTQ :
# R1 FASTQ situé dans un bucket S3 us-east-1
s3://1000genomes/phase3/data/NA12878/sequence_read/ERR001268_1.filt.fastq.gz
# R2 FASTQ situé dans un bucket S3 us-east-1
s3://1000genomes/phase3/data/NA12878/sequence_read/ERR001268_2.filt.fastq.gz# Exemple de commande pour copier un objet :
YOUR_FASTQ_BUCKET=<BUCKET_NAME_HERE># Pour AWS, utilisez :
aws s3 cp s3://1000genomes/phase3/data/NA12878/sequence_read/ERR001268_1.filt.fastq.gz s3://{YOUR_FASTQ_BUCKET}/FASTQ/
# Pour GCP, copiez le fichier vers le stockage local d'une VM avec 'aws s3 cp'
# Puis, copiez-le du stockage local vers un bucket GCS avec gsutil :
gsutil -o GSUtil:parallel_composite_upload_threshold=150M cp /tmp/FASTQ/ERR001268_1.filt.fastq.gz gs://{YOUR_FASTQ_BUCKET}/FASTQ/
Vous devrez également récupérer le dernier génome humain de référence et construire un index bwa-mem2 associé. Je recommande de :
- Copier le FASTA hg38 depuis le bucket S3 us-east-1 du Broad Institute vers une VM à très haute mémoire dans le cloud de votre choix : par exemple une r5.16xlarge sur AWS ou une n2-highmem-64 sur GCP.
- Installer bwa-mem2 (ou utiliser l'image de conteneur décrite plus loin)
- Construire l'index de référence bwa-mem2 (compter 1 à 2 heures)
- Téléverser les fichiers de sortie dans un nouveau bucket de votre compte cloud
# Génome de référence hg38 du Broad Institute situé dans un bucket S3 us-east-1
s3://broad-references/hg38/v0/Homo_sapiens_assembly38.fasta# Exemple de commande pour construire l'index bwa-mem2 et le sauvegarder dans votre bucket
YOUR_REF_BUCKET=<BUCKET_NAME_HERE># Pour AWS et GCP, utilisez :
aws s3 cp s3://broad-references/hg38/v0/Homo_sapiens_assembly38.fasta /tmp/reference/hg38.fastabwa-mem2 index -p hg38 /tmp/reference/hg38.fasta# Pour AWS, utilisez :
aws s3 sync /tmp/reference/ s3://${YOUR_REF_BUCKET}/hg38/
# Pour GCP, utilisez :
gsutil -m -o GSUtil:parallel_composite_upload_threshold=150M cp "/tmp/reference/hg38*" gs://${YOUR_REF_BUCKET}/hg38/
Si vous avez correctement suivi les étapes ci-dessus, vous disposez désormais de deux FASTQ (données brutes de séquençage du génome) et du génome humain de référence hg38, prêts à servir aux tests de workflow bioinformatique.
Étape 2 : Construire votre infrastructure cloud
Avec Terraform, nous allons provisionner toute l'infrastructure cloud nécessaire au fonctionnement d'un cluster Kubernetes entièrement managé, et exécuter quelques commandes sur le cluster (via kubectl) pour y installer argo, un outil Kubernetes dédié à l'exécution de workflows (par exemple, des pipelines bioinformatiques).
Par défaut, Terraform conserve l'état de l'infrastructure cloud en local. En conditions réelles, on utilise plutôt un bucket comme backend Terraform : ainsi, lorsqu'un collaborateur modifie l'infrastructure, tout le monde s'appuie sur la même source de vérité, à jour.
Un bucket en backend verrouille également les modifications d'infrastructure tant qu'un changement est en cours d'application, ce qui évite l'exécution simultanée d'opérations Terraform potentiellement contradictoires. Pour activer cette prise en charge, créez un bucket dans votre environnement cloud, puis remplacez toutes les références à doit-matt-tf-state-test dans la base de code par le nom de votre bucket.
Une fois ce bucket de backend Terraform prêt, vous pouvez provisionner l'infrastructure.
Pour exécuter votre workload sur Google Cloud, placez-vous dans GCP/terraform. Ce dossier contient les sous-dossiers standard et autopilot, qui regroupent chacun le code TF permettant de lancer un cluster GKE en mode Standard ou Autopilot. Autopilot est généralement à privilégier : il rapproche les nœuds (ressources de calcul) d'un modèle serverless tout en imposant de nombreuses bonnes pratiques de sécurité.
Malheureusement, Autopilot ne fonctionnera pas dans cette démo en raison d'une limitation : l'espace disque éphémère est plafonné à 10 Gio, ce qui ne suffit pas pour exécuter FastQC ni bwa-mem2 sur des données réelles. Si cette limitation venait à être levée, l'usage d'Autopilot plutôt que de Standard serait toutefois recommandé. J'ai prévu du code TF pour Autopilot au cas où cette limite serait relevée après publication. AWS ECS Fargate présente lui aussi une limite d'espace disque de 200 Gio, insuffisante pour la plupart des usages réels en calcul scientifique. Le code TF Fargate n'est pas couvert dans le dépôt.
Pour exécuter votre workload sur AWS, placez-vous dans AWS/terraform.
Une fois dans le dossier terraform propre à votre cloud, exécutez :
terraform apply -auto-approve# Lancez d'abord 'terraform plan' pour voir la liste des modifications d'infrastructure planifiées sans rien créer
Cette commande planifie puis exécute immédiatement l'ensemble des fichiers terraform '.tf' présents dans le dossier afin de provisionner un cluster K8s et ses ressources prérequises. Terraform vous demandera un ID de projet GCP ou un ID de compte AWS, et c'est tout.
Avant le apply, vous pouvez éventuellement modifier le fichier variables.tf pour changer la région de lancement du cluster, son nom, coder en dur la variable d'ID de projet GCP/compte AWS, etc.
Je vous encourage vivement à étudier le code pour vous familiariser avec Terraform
Pour vous aider à prendre vos repères, voici ce qui est créé pour GCP :
- Activation des API Compute, Container et IAM
- Un cluster GKE Standard avec auto-scaling au sein du VPC par défaut, utilisant la version Kubernetes par défaut et à jour (v1.20 au moment de la publication), avec quelques fonctionnalités de sécurité optionnelles activées : Workload Identity, Shielded Nodes et Secure Boot. C'est un cluster régional, avec des nœuds répartis sur plusieurs zones (centres de données)
- Un compte de service GCP (SA) et son compte de service Kubernetes lié (KSA), qui permettent à Workload Identity de restreindre le périmètre des permissions GCP pour tous les jobs exécutés dans un namespace Kubernetes donné. Ce SA/KSA pourra lire/écrire dans les buckets, et envoyer divers logs et métriques à Cloud Logging et Cloud Metrics
- Le cluster s'appuie sur des instances préemptibles / Spot. Vous paierez nettement moins que le tarif à la demande, en contrepartie de nœuds susceptibles de disparaître brusquement et d'être remplacés. K8s identifie automatiquement les jobs en échec dus à une coupure soudaine de nœud et les replanifie
Pour AWS, les composants réseau et sécurité suivants sont créés :
- Un nouveau VPC dédié au cluster K8s
- Des sous-réseaux publics (inutilisés) et privés, ainsi que les tables de routage pour les nœuds du cluster
- Une NAT Gateway, des endpoints VPC S3 et ECR avec leurs security groups associés activant TLS, et l'activation du support des noms d'hôtes DNS pour permettre un cluster à nœuds privés
- Une Internet Gateway permettant l'accès public au plan de contrôle du cluster
Pour AWS, vous devrez également vous placer dans AWS/terraform/ecr et exécuter à nouveau terraform apply, car les fichiers .tf de ce dossier créent :
- Les dépôts dans lesquels seront stockées les images de conteneurs que vous créerez
(Sur GCP, ces dépôts n'ont pas besoin d'être créés)
Pour AWS, vous devrez également vous placer dans AWS/terraform/eks et exécuter à nouveau terraform apply, car les fichiers .tf de ce dossier créent :
- Un rôle IAM pour le cluster, autorisant la lecture et l'écriture dans les buckets S3, ainsi qu'un accès SSH aux nœuds via le navigateur, sans clé, grâce à SSM Manager
- Un cluster EKS avec auto-scaling utilisant la version Kubernetes par défaut et à jour (v1.21 au moment de la publication). Workload Identity, Shielded Nodes et Secure Boot ne sont pas directement disponibles sur EKS. C'est un cluster régional, avec des nœuds répartis sur plusieurs zones (centres de données)
- Le cluster s'appuie sur des instances préemptibles / Spot. Vous paierez nettement moins que le tarif à la demande, en contrepartie de nœuds susceptibles de disparaître brusquement et d'être remplacés. K8s identifie automatiquement les jobs en échec dus à une coupure soudaine de nœud et les replanifie
Une fois la ou les opérations terraform apply terminées, votre cluster est opérationnel et prêt à recevoir des commandes de l'API Kubernetes via kubectl. Authentifiez simplement kubectl avec une commande propre à votre cloud :
# Pour GCP :
gcloud container clusters get-credentials --region=us-central1 bioinformatics-tasks
# Pour AWS :
aws eks update-kubeconfig --name=bioinformatics-tasks
Vous pouvez désormais exécuter diverses commandes sur le cluster, comme lister les pods déployés dans tous les namespaces avec kubectl get pods -A :
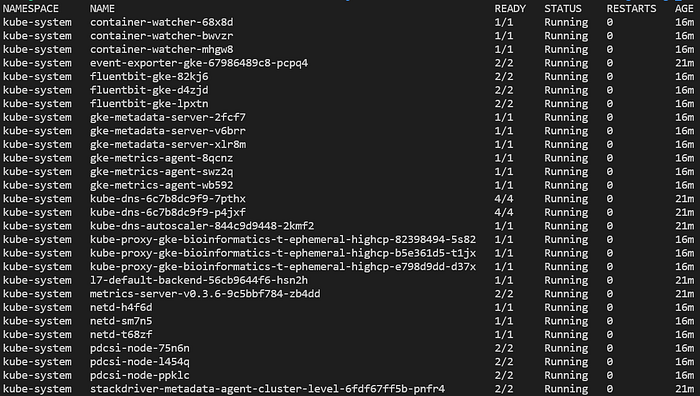
Pods typiques d'un nouveau cluster GKE Standard entièrement managé
Le cluster en place, il nous reste à y installer argo, qui permettra de soumettre non seulement des tâches individuelles, mais aussi un workflow / pipeline de tâches piloté par DAG.
Sur GCP, placez-vous dans GCP/terraform/standard/kubectl_commands
Sur AWS, placez-vous dans AWS/terraform/kubectl_commands
L'exécution de terraform apply -auto-approve dans ces dossiers déclenche des commandes kubectl pilotées par Terraform, qui :
- Créent un namespace 'biojobs' destiné à recevoir les jobs et (sur GCP) restreint en permissions à la lecture/écriture d'objets dans un bucket.
- Installent argo et, sur GCP via Workload Identity, lui accordent les permissions appropriées pour suivre la progression des pods dans le namespace 'biojobs'.
Vous pouvez vérifier la création du namespace et l'installation d'argo en listant tous les namespaces, puis en interrogeant les pods en cours d'exécution dans le namespace argo :
kubectl get ns
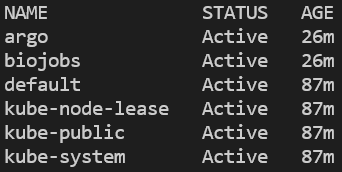
Tous les namespaces du cluster. Sur EKS, on retrouvera également 'amazon-cloudwatch'.
kubectl get pods -n argo
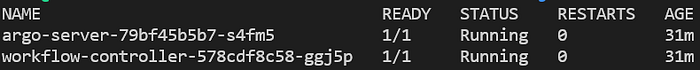
Pods du serveur Argo et du contrôleur de workflow, en cours d'exécution dans le namespace 'argo'
Enfin, exécutez cette commande pour vous assurer qu'argo s'accommode du passage de Kubernetes ≥v1.19 de docker à containerd comme runtime de conteneurs. Ne vous attardez pas sur ce détail : argo finira par corriger ce point :
kubectl patch configmap workflow-controller-configmap -n argo --patch ‘{"data":{"containerRuntimeExecutor":"k8sapi"}}’
Argo installé et nœuds de calcul prêts, place à la pratique !
Étape 3 : Construire les images de conteneurs FastQC et bwa-mem2
Pour exécuter les workloads FastQC et bwa-mem2 sur AWS ou GCP, nous devons construire des images de conteneurs pour ces outils et les stocker dans le dépôt d'images de conteneurs propre à chaque cloud (GCR pour GCP, ECR pour AWS).
Placez-vous dans images/fastqc/
Ce dossier contient le Dockerfile qui définit comment empaqueter FastQC dans un conteneur, accompagné de s5cmd (un outil pour des copies AWS S3 ←→ local rapides) et de gsutil (pour les copies GCS ←→ local). Prenez le temps d'en parcourir le contenu et de vous documenter sur la construction des Dockerfiles. À ce sujet, cet article que j'ai rédigé sur la maximisation des vitesses de transfert vers les buckets pourra vous intéresser : ses principes sont repris dans le Dockerfile.
Exécutez build_fastqc_image_gcp.sh ou build_fastqc_image_aws.sh pour créer une image de conteneur à partir du Dockerfile, puis la pousser respectivement vers GCR ou ECR.
Pour créer une image de conteneur dédiée à bwa-mem et la stocker dans GCR ou ECR, placez-vous dans images/bwa-mem2/ et reproduisez le même processus d'exécution du script de build.
Cette étape terminée, FastQC et bwa-mem2 sont disponibles pour exécution sur un cluster Kubernetes via les images de conteneurs stockées dans le dépôt d'images de votre environnement cloud.
Étape 4 : Soumettre et surveiller des jobs Kubernetes
Plaçons-nous maintenant dans le dossier de jobs Kubernetes propre à votre cloud pour en lancer quelques-uns. À terme, FastQC et bwa-mem2 s'enchaîneront dans un pipeline avec argo, mais pour démarrer, nous exécuterons ces tâches une à une.
Pour GCP, placez-vous dans GCP/gke/FastQC/
Pour AWS, placez-vous dans AWS/eks/FastQC/
Ces dossiers contiennent un Job Kubernetes nommé fastqc.yaml.
Ce YAML définit un job exécutable avec kubectl create -f fastqc.yaml. Le fichier contient toutefois quelques variables qui doivent être définies puis substituées par run_fastqc_job.sh. Prenez le temps de comprendre les objets Kubernetes en général et plus particulièrement les jobs pour décortiquer fastqc.yaml, et examinez aussi ce que fait le script shell.
En substance, Kubernetes sert le plus souvent à exécuter des pods de longue durée appelés Deployments (par exemple, des serveurs web persistants), mais il peut aussi exécuter des Jobs, c'est-à-dire des tâches d'exécution unique, comme des programmes scientifiques tels que FastQC. Tout au long de la base de code, nous soumettrons des objets Job Kubernetes au cluster.
Renseignez les variables INPUT_FILEPATH et OUTPUT_FILEPATH dans le script shell : il lancera alors un job FastQC sur l'un des FASTQ récupérés plus tôt, puis enregistrera le rapport compressé dans un bucket. Vous pouvez aussi remplacer manuellement les variables dans fastqc.yaml et exécuter le job avec : kubectl create -f fastqc.yaml
Suivez la progression de FastQC avec : kubectl get pods -n biojobs
Vous remarquerez qu'un seul pod est lancé : il commence par tirer l'image de conteneur, puis exécute peu après le job spécifié :

Pod FastQC — statut ContainerCreating

Pod FastQC — statut Running
Consultez les logs de l'application en cours d'exécution avec kubectl logs -n biojobs <pod_name> :
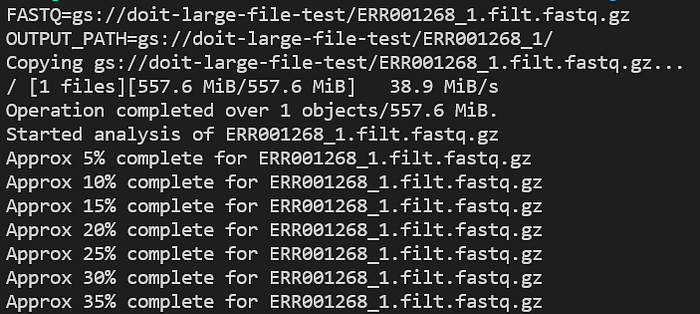
Progression du pod FastQC
Les conteneurs — tout comme GKE/EKS — étant pensés pour la supervision, vous retrouverez facilement ces entrées de logs dans la solution de logging native de votre cloud, ce qui vous donne une rétention longue durée et de vraies capacités d'investigation.
Recherchez l'expression complete for dans Cloud Logging de GCP ou CloudWatch d'AWS pour localiser les entrées de logs des pods FastQC :
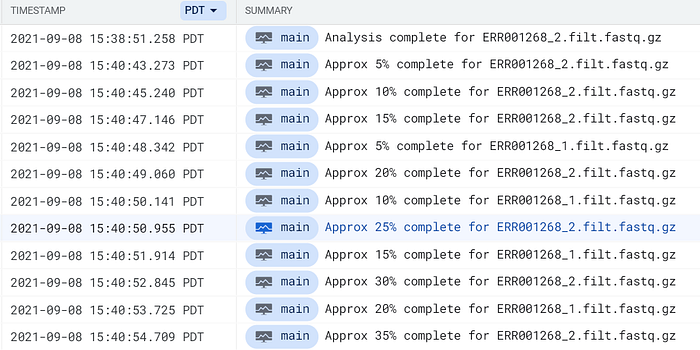
Logs des pods FastQC remontés dans Cloud Logging de GCP
Vous devriez finir par voir un rapport FastQC apparaître dans le chemin de votre bucket de sortie. Lancez des pods FastQC pour les deux fichiers FASTQ jusqu'à voir leurs rapports respectifs :
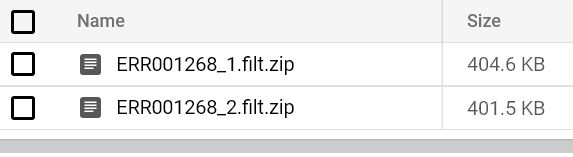
Rapports FastQC stockés dans le bucket
Voilà tout ce qu'il faut pour exécuter FastQC à grande échelle sur Kubernetes !
Que vous lanciez un seul job FastQC ou dix mille en simultané, le plan de contrôle Kubernetes, conjugué au service d'auto-scaling des nœuds de calcul de l'environnement cloud, ajustera la capacité de calcul à la hausse comme à la baisse pour répondre à la demande, conformément aux exigences cpu/mémoire/disque définies dans le YAML de l'objet Kubernetes FastQC.
Veillez simplement à fixer, dès la création du cluster, des valeurs maximales suffisantes pour l'autoscaler en termes de nombre de nœuds / cpu / mémoire — ou au moins au niveau souhaité pour maîtriser les coûts. (GCP : gke.tf AWS : cluster.tf)
Vous pouvez exécuter bwa-mem2 à grande échelle de manière tout aussi simple et rentable !
Pour GCP, placez-vous dans GCP/gke/bwa-mem2/
Pour AWS, placez-vous dans AWS/eks/bwa-mem2/
Renseignez les chemins d'entrée/sortie dans les variables en haut de run_bwa-mem2_job.sh : elles remplaceront les noms de variables dans bwa-mem2.yaml, puis le Job sera soumis à votre cluster.
Continuez de surveiller le statut et les logs des pods jusqu'à voir un fichier BAM apparaître dans le chemin de votre bucket de sortie :
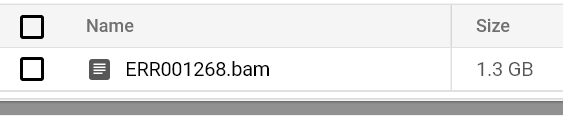
Fichier BAM stocké dans le bucket
En consultant les métriques d'utilisation CPU des nœuds GCE / EC2 qui alimentent votre cluster, vous remarquerez que les jobs FastQC mono-cœur, peu intensifs, sont envoyés sur des machines à faible CPU, tandis que les jobs BWA, qui exploitent tous les cœurs, sont dirigés vers des machines à fort CPU. Cela correspond aux groupes de nœuds Kubernetes faible/fort CPU créés pour s'assurer que des machines bien dimensionnées soient affectées à chaque type de workload, avec une montée et descente en charge automatique et économique.
Sur AWS, par exemple, il n'aurait aucun sens de provisionner une m5.24xlarge à 96 vCPU pour exécuter un simple nouveau job FastQC mono-cœur ; on soumet donc FastQC à un groupe de nœuds composé de m5.xlarge à 4 vCPU. À l'inverse, bwa tire profit de chaque cœur disponible : ces jobs sont donc dirigés vers le groupe de nœuds à 96 vCPU, qui s'ajuste indépendamment du groupe à 4 vCPU (FastQC).
Si vous avez déjà construit des workflows d'analyse secondaire qui prennent des données brutes de séquençage et les traitent pour découvrir des variants, vous commencez sans doute à mesurer toute la puissance de Kubernetes. Fini :
- Les schedulers de jobs on-prem aux performances capricieuses à grande échelle
- L'angoisse de savoir si lancer trop de jobs en même temps va faire tomber le scheduler, voire le cluster lui-même qui alimente vos workloads (et ceux de tout le monde).
- Les frictions avec les collègues lorsque vous monopolisez la capacité de calcul on-prem au détriment de leurs propres workloads.
- La surveillance et la relance manuelle de workloads en échec pour des raisons imprévues mais récupérables.
- Les heures perdues à tenter de retrouver les fichiers de log de jobs plantés — souvent dans des applications dont la fonctionnalité de logging est mal pensée.
Reste maintenant à éviter d'écrire du code spaghetti pour enchaîner les étapes…
Étape 5 : Soumettre des workflows Argo (jobs Kubernetes pipelinés)
Argo permet d'enchaîner des jobs en pipeline tout en restant opérationnel à grande échelle, et prend en charge les nouvelles tentatives, aussi bien au niveau des jobs que des pipelines, en cas de défaillance imprévue.
Voyons comment utiliser argo pour exécuter deux pods FastQC en parallèle. Si — et seulement si — ces deux pods se terminent avec succès, l'alignement avec bwa-mem2 sera lancé sur la même paire FASTQ.
(Exercice personnel : vous pourriez ajouter une étape pass/no-pass à l'image FastQC, de sorte que si le FASTQ est de qualité trop faible pour justifier un alignement, l'étape BWA du workflow argo soit ignorée.)
Pour GCP, placez-vous dans GCP/gke/argo_workflow/
Pour AWS, placez-vous dans AWS/eks/argo_workflow/
Renseignez les chemins d'entrée/sortie dans les variables en haut de run_fastqc_to_bwa.sh : elles remplaceront les noms de variables dans fastqc_to_bwa.yaml, puis le Job sera soumis à votre cluster.
Lancez en continu kubectl get pods -n biojobs : vous verrez les pods FastQC s'exécuter, puis se terminer, suivis du lancement d'un job BWA :

Pods FastQC en cours d'exécution sur les fichiers FASTQ R1 et R2 de NA12878
…
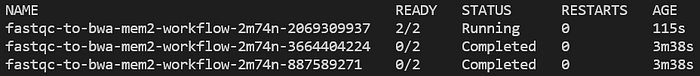
Pod d'alignement bwa-mem2 lancé après la fin des deux pods FastQC
Examinons le pod bwa-mem2 avec :
kubectl logs -n biojobs <pod_name> main
(Notez l'ajout de 'main' après le nom du pod : avec argo, chaque job d'un workflow comporte des logs de pods 'main' et 'wait'.)
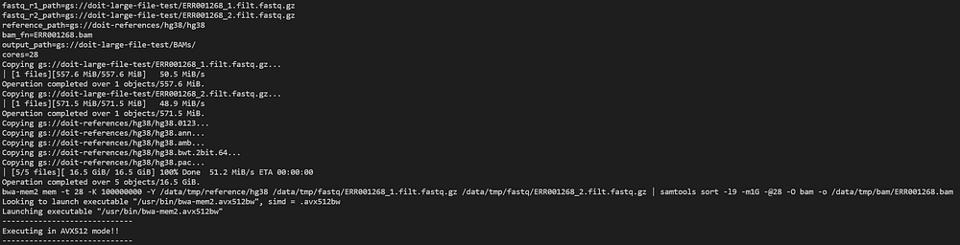
bwa-mem2 en plein travail au sein d'un workflow argo
Exécutez kubectl get wf -n biojobs pour suivre la progression du workflow argo :

À la fin de BWA, le workflow sera terminé et vous trouverez une paire de rapports FastQC ainsi qu'un BAM dans votre bucket de sortie.
Étape 6 : Nettoyage
Nous avons provisionné une infrastructure de calcul plutôt onéreuse pour faire tourner BWA ; pas question de laisser ce cluster GKE/EKS actif lorsqu'il n'est pas utilisé.
Parcourez simplement les dossiers Terraform en sens inverse et exécutez :
terraform destroy -auto-approve
Notez que cela ne supprimera pas les buckets que vous avez créés manuellement pour servir de backend Terraform et stocker les fichiers FASTQ et de référence. Terraform ne connaît et ne sait surveiller que les ressources qu'il a lui-même provisionnées. Sur GCP, vous devrez aussi supprimer manuellement les images de conteneurs dans GCR.
Sur GCP, exécutez la commande destroy dans cet ordre :
GCP/terraform/standard/kubectl_commands/
GCP/terraform/standard/
Sur AWS, exécutez la commande destroy dans cet ordre :
AWS/terraform/eks/kubectl_commands/
AWS/terraform/eks/
AWS/terraform/ecr/ (si vous souhaitez supprimer les dépôts d'images)
AWS/terraform/
Étape 7 : Et après ?
Avec les compétences acquises jusqu'ici, vous pouvez construire des pipelines d'une complexité arbitraire reposant sur n'importe quelle application, puisque tout programme peut être conteneurisé.
Avec un code d'image de conteneur versionné, des conteneurs taggés par version, une infrastructure et des pipelines argo versionnés, vous pouvez déployer de nouveaux logiciels et de nouvelles versions de pipelines, déprécier les anciennes versions et faire un rollback en cas de déploiement défectueux, avec une relative facilité — du moins comparé aux solutions alternatives — pour peu que vous poursuiviez ce parcours d'apprentissage.
À vous de définir les prochaines étapes, mais j'espère vous avoir transmis les compétences fondamentales du calcul scientifique moderne et scalable.
Dans le cloud, les bonnes pratiques ne suffisent qu'à un certain point. Concevoir pour la haute disponibilité, la résilience aux pannes et une scalabilité optimisée en coûts est extrêmement difficile aujourd'hui. Ces tâches d'ingénierie supplémentaires viennent s'ajouter à une pile de workloads en perpétuelle croissance, ce qui pèse directement sur la productivité.
En tant que scientifique du XXIe siècle, il est essentiel de concevoir des applications avec la scalabilité et la facilité de déploiement en tête. Faire l'impasse, c'est créer des casse-têtes pour votre organisation. À long terme, ces difficultés finiront par dégénérer en problèmes de production sérieux et sans fin.
Si ce projet vous inspire, mais qu'il vous semble aussi un peu intimidant, n'hésitez pas à contacter DoiT International . Vous pouvez également m'écrire sur LinkedIn .
DoiT met un point d'honneur à accompagner ses clients vers des architectures cloud optimisées en coûts, scalables, hautement disponibles, résilientes et taillées pour le big data — un service offert littéralement gratuitement. Nous avons reçu le titre de Global Reseller Partner of the Year 2020 de Google Cloud, et ce n'est pas un hasard.
Nous avons obtenu la Distinction de Certification Partner Network (APN) d'Amazon tout en conservant le statut d'AWS Advanced Consulting Partner, grâce à des talents d'ingénierie de premier plan et à un solide support client. Toutes les informations sont communiquées à la demande pour répondre à vos questions cloud ou résoudre les problèmes en cours.
Merci beaucoup pour votre temps. J'espère que ces deux articles vous auront aidé à enrichir vos compétences. Vous montez en puissance ? Contactez-nous, nous vous aiderons à atteindre vos objectifs.
Merci de votre lecture ! Pour rester en contact, suivez-nous sur le DoiT Engineering Blog , le DoiT Linkedin Channel et le DoiT Twitter Channel . Pour explorer les opportunités de carrière, rendez-vous sur https://careers.doit-intl.com .