Boas-vindas à Parte 2 de Computação Científica em Nuvem em Escala Global com Kubernetes e Terraform. Aqui eu compartilho uma demonstração completa de computação científica em nuvem em escala global com Kubernetes e Terraform. Se quiser relembrar ou se aprofundar nos princípios e conceitos modernos de DevOps, dê uma olhada na Parte 1 desta série.
Este guia mostra como DevOps, Containers, Kubernetes e Terraform podem ser usados em implantações reais de workloads de computação.
PerfectScale™ for Kubernetes
Ready to optimize?
Get your free Kubernetes savings analysis
Visão geral
De forma resumida, o código de exemplo:
- Mostra como empacotar ferramentas comuns de bioinformática (FastQC e BWA) em imagens de container e enviá-las para o repositório de imagens de cada nuvem
- Provisiona (e permite remover rapidamente) toda a infraestrutura de nuvem necessária para executar workloads via Terraform, uma ferramenta de Infraestrutura como Código
- Implanta um pipeline de workflow comum baseado nessas imagens e na infraestrutura provisionada no serviço totalmente gerenciado de Kubernetes de cada nuvem, um sistema de gerenciamento e orquestração de execução de containers. O Argo será usado para orquestrar um workflow ou pipeline de tarefas em um cluster Kubernetes
Combinando Docker, Terraform, Kubernetes e Argo, você vai aprender a:
- Provisionar, atualizar e remover a infraestrutura necessária para rodar seus workloads com comandos simples
- Executar workloads orientados por DAG de ponta a ponta com novas tentativas automáticas em etapas individuais quando houver erros inesperados ou falhas de infraestrutura
- Centralizar logs e monitoramento de métricas dos workloads em ferramentas nativas da nuvem para exploração de logs e métricas
- Rodar workloads em uma infraestrutura que escala automaticamente para cima (até a capacidade em escala global) e para baixo (poucos ou nenhum recurso de computação) conforme a necessidade, de modo que você só pague pelos recursos de CPU/RAM/armazenamento usados para concluir suas tarefas. Acabou a era de deixar servidores ociosos rodando e inflando sua fatura à toa
- Implantar novas versões de software de forma transparente e descontinuar versões antigas
Your cloud bill shouldn't be a mystery
Optimization, automation, expertise. In one platform.
Passo a passo do código
Com um entendimento básico de DevOps em mãos, vamos ver como a base de código que acompanha este post demonstra os princípios fundamentais de DevOps. Este passo a passo mostra como construir a infraestrutura para sustentar a execução de pipelines científicos com capacidade de escala global. Mais especificamente, vamos rodar componentes de um pipeline comum de bioinformática.
Queremos:
- Gerar relatórios sobre um par de arquivos brutos de sequenciamento de genoma para validar que são de alta qualidade. Os dados brutos de sequenciamento são armazenados no formato FASTQ, e as verificações de QC são feitas com uma ferramenta chamada FastQC.
- Depois desses relatórios, executar o processo conhecido como alinhamento, que, em última instância (com processamento adicional por ferramentas downstream), permite a descoberta de mutações. Isso é feito com uma ferramenta chamada bwa-mem2.
Ou seja, queremos que nosso pipeline baseado em Kubernetes execute simultaneamente dois containers FastQC sobre um par de arquivos FASTQ de sequenciamento e, se as duas tarefas forem bem-sucedidas, rode o alinhamento bwa-mem2 do par de datasets brutos contra a versão mais recente do genoma humano, conhecida como hg38. Tanto os relatórios FastQC quanto o arquivo de alinhamento gerado pelo BWA, conhecido como arquivo BAM, serão enviados a um bucket para armazenamento de longo prazo.
Atenção ao custo
Como esta base de código vai te guiar por um caso de uso analítico real, e não por um exemplo simples, é esperado que o ambiente de computação configurado tenha um custo nada desprezível. Dependendo de quanto tempo você deixar a infraestrutura do Terraform rodando, em geral espere gastar valores na casa das dezenas de dólares na sua fatura de nuvem.
Passo 0: Instale os clientes locais: CLI da nuvem, Terraform e kubectl
Garanta que a CLI da sua nuvem esteja instalada localmente e autenticada na conta que você vai usar. Links das CLIs: GCP e AWS. Provisionar infraestrutura como código na nuvem é tão simples quanto entrar em uma pasta com arquivos YAML do Terraform e executar:
terraform apply -auto-approve
Para isso funcionar, você precisa instalar o cliente terraform localmente. O Terraform vai usar automaticamente as credenciais de autenticação da CLI da sua nuvem.
Executar um job ou workflow no Kubernetes é tão simples quanto criar o template YAML para esse job/workflow e rodar:
kubectl create -f job.yaml
Para isso funcionar, você precisa instalar o cliente kubectl localmente. O kubectl será autenticado com um comando específico de cada CLI de nuvem.
Passo 1: Obter os dados FASTQ e a Referência Humana (hg38)
Antes de começar a demonstração do workflow, você vai precisar de um par de arquivos FASTQ de exemplo e do genoma humano de referência hg38.
Para manter os custos de processamento ao mínimo e ainda usar dados em escala de caso de uso real, baixei dois arquivos FASTQ de cerca de 0,5 GB para a amostra ‘gold standard’ NA12878 do bucket S3 do projeto 1000 Genomes. Recomendo bastante criar um bucket GCS ou S3 na sua conta de nuvem e copiar esses arquivos para lá. Como você vai testar o workflow várias vezes, é melhor não baixar repetidamente do bucket S3 do 1000G.
Fique sempre atento ao preço de transferência de dados entre regiões .
Os arquivos FASTQ estão localizados na região us-east-1 da Amazon, então mover esses dados para outro bucket S3 regional (por exemplo, a região us-west-2 que usei nos testes) ou para fora da AWS, em direção a um bucket Cloud Storage do GCP, terá um custo por cada GB transferido para fora da região do bucket. Crie um bucket na região de nuvem desejada e copie os FASTQs para ele:
# R1 FASTQ em um bucket S3 us-east-1
s3://1000genomes/phase3/data/NA12878/sequence_read/ERR001268_1.filt.fastq.gz
# R2 FASTQ em um bucket S3 us-east-1
s3://1000genomes/phase3/data/NA12878/sequence_read/ERR001268_2.filt.fastq.gz# Comando de exemplo para copiar um objeto:
YOUR_FASTQ_BUCKET=<BUCKET_NAME_HERE># Para AWS, use:
aws s3 cp s3://1000genomes/phase3/data/NA12878/sequence_read/ERR001268_1.filt.fastq.gz s3://{YOUR_FASTQ_BUCKET}/FASTQ/
# Para GCP, copie o arquivo para o armazenamento local de uma VM com 'aws s3 cp'
# Depois, copie do armazenamento local para um bucket GCS com gsutil:
gsutil -o GSUtil:parallel_composite_upload_threshold=150M cp /tmp/FASTQ/ERR001268_1.filt.fastq.gz gs://{YOUR_FASTQ_BUCKET}/FASTQ/
Você também vai precisar pegar o genoma humano de referência mais recente e construir um índice bwa-mem2 para ele. Recomendo:
- Copiar o FASTA hg38 do bucket S3 us-east-1 do Broad Institute para uma VM com altíssima memória na nuvem de sua escolha. Pode ser uma máquina r5.16xlarge na AWS ou uma n2-highmem-64 no GCP.
- Instalar o bwa-mem2 (ou usar a imagem de container descrita mais adiante)
- Construir o índice de referência bwa-mem2 (leva de 1 a 2 horas)
- Enviar os arquivos de saída para um novo bucket na sua conta de nuvem
# Genoma humano de referência hg38 do Broad Institute, em um bucket S3 us-east-1
s3://broad-references/hg38/v0/Homo_sapiens_assembly38.fasta# Comando de exemplo para construir o índice bwa-mem2 e salvá-lo no seu bucket
YOUR_REF_BUCKET=<BUCKET_NAME_HERE># Para AWS e GCP, use:
aws s3 cp s3://broad-references/hg38/v0/Homo_sapiens_assembly38.fasta /tmp/reference/hg38.fastabwa-mem2 index -p hg38 /tmp/reference/hg38.fasta# Para AWS, use:
aws s3 sync /tmp/reference/ s3://${YOUR_REF_BUCKET}/hg38/
# Para GCP, use:
gsutil -m -o GSUtil:parallel_composite_upload_threshold=150M cp "/tmp/reference/hg38*" gs://${YOUR_REF_BUCKET}/hg38/
Se seguiu corretamente os passos acima, você agora terá dois FASTQs (dados brutos de sequenciamento de genoma) e o genoma humano de referência hg38 prontos para usar nos testes do workflow de bioinformática.
Passo 2: Construa sua infraestrutura de nuvem
Com o Terraform, vamos provisionar toda a infraestrutura de nuvem necessária para rodar um cluster Kubernetes totalmente gerenciado e executar alguns comandos no cluster (via kubectl) que instalam o argo — uma ferramenta Kubernetes para executar workflows (por exemplo, pipelines de bioinformática).
Por padrão, o Terraform mantém o estado conhecido da infraestrutura de nuvem localmente. Em casos de uso reais, no entanto, você usa um bucket como backend do Terraform, para que, quando alguém da empresa fizer uma mudança de infraestrutura, todos operem a partir do mesmo dataset atualizado de ‘verdade’ sobre o estado dessa infraestrutura.
Usar um bucket como backend também bloqueia mudanças de infraestrutura enquanto outro conjunto de mudanças ainda está sendo aplicado, evitando que múltiplas operações de apply do terraform — potencialmente conflitantes — sejam executadas ao mesmo tempo. Para adicionar suporte a um backend Terraform baseado em bucket, crie um bucket no seu ambiente de nuvem e, depois, faça uma busca e substituição em toda a base de código de doit-matt-tf-state-test pelo nome do seu bucket.
Com um bucket de backend Terraform pronto, dá para provisionar a infraestrutura.
Se quiser rodar seu workload no Google Cloud, vá até GCP/terraform. Dentro dessa pasta, há subpastas chamadas standard e autopilot, cada uma com código TF que vai criar um cluster GKE no modo Standard ou Autopilot. O Autopilot costuma ser a opção preferida, pois não só leva os nós (recursos de computação) a um modelo serverless, como também impõe o uso de várias melhores práticas de segurança.
Infelizmente, o Autopilot não vai funcionar nesta demo por causa de uma limitação: o espaço de disco efêmero é limitado a 10 GiB, o que não é suficiente para rodar FastQC nem bwa-mem2 com dados reais. Mesmo assim, caso essa limitação seja removida no futuro, o uso do Autopilot em vez do Standard é incentivado. Tenho código TF para Autopilot caso essa limitação seja flexibilizada após a publicação. O AWS ECS Fargate também tem uma limitação de 200 GiB de espaço em disco, que não é suficiente para a maioria dos casos reais de computação científica. Código TF para Fargate não está coberto no repositório.
Se quiser rodar seu workload na AWS, vá até AWS/terraform.
Já dentro da pasta terraform específica da sua nuvem, execute:
terraform apply -auto-approve# Rode 'terraform plan' primeiro para ver a lista de mudanças planejadas sem criar nada
Isso vai planejar e, em seguida, executar imediatamente o conjunto de arquivos ‘.tf’ do terraform presentes na pasta para provisionar um cluster K8s e seus recursos pré-requisitos. O Terraform vai pedir um ID de Projeto GCP ou um ID de Conta AWS, e é só isso.
Opcionalmente, dá para editar o arquivo variables.tf antes de rodar o apply, caso queira mudar a região onde o cluster será criado, o nome do cluster, deixar a variável de projeto GCP/conta AWS hardcoded etc.
Recomendo bastante revisar o código para aprender Terraform
Para acelerar seu entendimento, os seguintes recursos são criados no GCP:
- Habilitação das APIs de Compute, Container e IAM
- Um cluster GKE Standard com auto-escalonamento dentro da VPC padrão usando a versão padrão e atualizada do Kubernetes (v1.20 no momento da publicação) com alguns recursos opcionais de segurança em melhores práticas habilitados, como Workload Identity, Shielded Nodes e Secure Boot. É um cluster regional, com nós distribuídos por múltiplas zonas (data centers)
- Uma Service Account (SA) do GCP e a Kubernetes Service Account (KSA) vinculada que habilitam o Workload Identity para limitar o escopo de permissão do GCP em todos os jobs executados dentro de um determinado namespace Kubernetes. A SA/KSA terá permissão para ler/escrever em buckets e gravar diversos logs e métricas no Cloud Logging e no Cloud Metrics
- O cluster usa instâncias preemptíveis / Spot. Você paga bem menos do que o preço sob demanda, com a possibilidade de os nós caírem inesperadamente e serem substituídos depois. O K8s identifica automaticamente os jobs que falharam por término súbito de nó e os reagenda
Para a AWS, os seguintes componentes de rede e segurança são criados:
- Uma nova VPC dedicada a hospedar o cluster K8s
- Sub-redes públicas (não usadas) e privadas + tabelas de rotas para os nós do cluster
- Um NAT Gateway, endpoints VPC para S3 e ECR e seus security groups habilitando TLS, além da habilitação do suporte a hostname DNS para sustentar um cluster de nós privado
- Um Internet Gateway para permitir acesso público ao plano de controle do cluster
Para a AWS, você também vai precisar ir até AWS/terraform/ecr e rodar terraform apply de novo, pois os arquivos .tf desta pasta criam:
- Os repositórios onde as imagens de container que você criar serão armazenadas
(Os repositórios não precisam ser criados no GCP)
Para a AWS, você também vai precisar ir até AWS/terraform/eks e rodar terraform apply de novo, pois os arquivos .tf desta pasta criam:
- Uma role IAM para o cluster habilitando acesso de leitura e escrita a buckets S3, além de SSH baseado em navegador, sem chave, nos nós via SSM Manager
- Um cluster EKS com auto-escalonamento usando a versão padrão e atualizada do Kubernetes (v1.21 no momento da publicação). Workload Identity, Shielded Nodes e Secure Boot não são recursos prontamente disponíveis no EKS. É um cluster regional, com nós distribuídos por múltiplas zonas (data centers)
- O cluster usa instâncias preemptíveis / Spot. Você paga bem menos do que o preço sob demanda, com a possibilidade de os nós caírem inesperadamente e serem substituídos depois. O K8s identifica automaticamente os jobs que falharam por término súbito de nó e os reagenda
Concluídas as operações de terraform apply, o cluster está operacional e pronto para receber comandos da API Kubernetes via kubectl. Basta autenticar o kubectl com um comando específico da nuvem:
# Para GCP:
gcloud container clusters get-credentials --region=us-central1 bioinformatics-tasks
# Para AWS:
aws eks update-kubeconfig --name=bioinformatics-tasks
Agora dá para rodar diversos comandos no cluster, como listar os pods implantados em todos os namespaces com kubectl get pods -A:
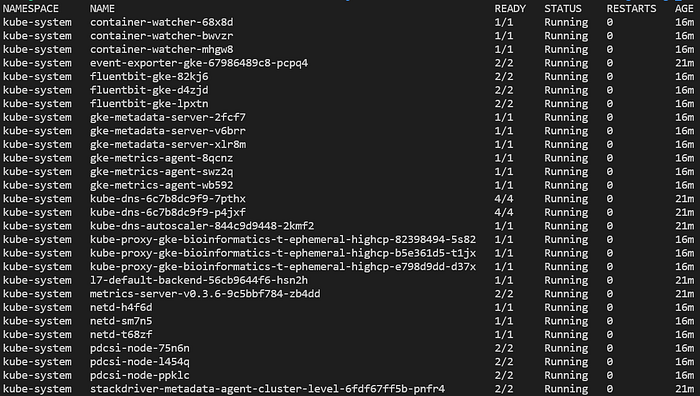
Pods típicos vistos em um novo cluster GKE Standard totalmente gerenciado
Com o cluster provisionado, agora precisamos instalar o argo nele, pois é o argo que vai permitir enviar não apenas tarefas individuais, mas um workflow / pipeline de tarefas direcionado por DAG.
Para fazer isso no GCP, vá até GCP/terraform/standard/kubectl_commands
Para fazer isso na AWS, vá até AWS/terraform/kubectl_commands
Executar terraform apply -auto-approve nessas pastas vai rodar comandos kubectl gerenciados pelo Terraform que:
- Criam um namespace ‘biojobs’ ao qual os jobs serão enviados e (no GCP) com escopo de permissões limitado a apenas ler/escrever objetos de um bucket.
- Instalam o argo e, no GCP com Workload Identity, dão a ele permissões com escopo apropriado para monitorar o progresso dos pods no namespace ‘biojobs’.
Você pode verificar se o namespace foi criado e o argo instalado listando todos os namespaces e, depois, consultando os pods em execução no namespace argo:
kubectl get ns
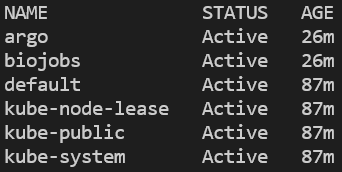
Todos os namespaces no cluster. O EKS também vai ter ‘amazon-cloudwatch.’
kubectl get pods -n argo
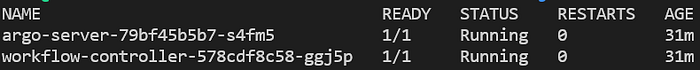
Pods do argo server e do workflow controller rodando no namespace ‘argo’
Por fim, rode este comando para garantir que o argo funcione bem com a mudança no Kubernetes ≥v1.19, do uso do docker como runtime de container para o containerd. Não se preocupe com esse detalhe, pois o argo vai resolver isso eventualmente:
kubectl patch configmap workflow-controller-configmap -n argo --patch ‘{"data":{"containerRuntimeExecutor":"k8sapi"}}’
Com o argo instalado e os nós de computação prontos, vamos colocar a mão na massa!
Passo 3: Construa as imagens de container do FastQC e do bwa-mem2
Para rodar workloads de FastQC e bwa-mem2 na AWS ou no GCP, vamos precisar construir imagens de container para essas ferramentas e armazená-las no respectivo repositório de imagens da sua nuvem (GCR para GCP, ECR para AWS).
Vá até images/fastqc/
Dentro desta pasta está o Dockerfile que define como empacotar o FastQC em um container junto com o s5cmd (ferramenta para cópias rápidas entre AWS S3 ↔ local) e o gsutil (para cópias entre GCS ↔ local). Reserve um tempo para revisar o conteúdo do arquivo e ler sobre como os Dockerfiles são construídos. A propósito, talvez você se interesse por este artigo que escrevi sobre como maximizar a velocidade de transferência de dados em buckets, já que os princípios desse artigo estão presentes no Dockerfile.
Execute build_fastqc_image_gcp.sh ou build_fastqc_image_aws.sh para criar uma imagem de container a partir do Dockerfile e, depois, enviá-la para o GCR ou ECR, respectivamente.
Para criar uma imagem de container para o bwa-mem e armazená-la no GCR ou ECR, vá até images/bwa-mem2/ e repita o mesmo processo de execução do script de build.
Com este passo concluído, você agora tem FastQC e bwa-mem2 disponíveis para execução em um cluster Kubernetes via as imagens de container armazenadas no repositório de imagens do seu ambiente de nuvem.
Passo 4: Envie e monitore jobs Kubernetes
Em seguida, vamos até a pasta específica da sua nuvem para jobs Kubernetes e lançar alguns. Em algum momento rodaremos FastQC e bwa-mem2 em um pipeline com o argo, mas, para começar, vamos rodar essas tarefas individualmente.
Para GCP, vá até GCP/gke/FastQC/
Para AWS, vá até AWS/eks/FastQC/
Dentro destas pastas há um Job Kubernetes chamado fastqc.yaml.
Este YAML define um job que pode ser executado com kubectl create -f fastqc.yaml. Mas o arquivo contém algumas variáveis que devem ser definidas e substituídas por run_fastqc_job.sh. Reserve um tempo para entender os objects do Kubernetes em geral e os jobs em específico para decifrar o fastqc.yaml, e também revise o que está acontecendo no shell script.
Em essência, o Kubernetes é mais usado para rodar pods de longa duração chamados Deployments (por exemplo, servidores web de longa duração), mas também pode rodar Jobs, ou tarefas de execução única, como programas científicos do tipo FastQC. Ao longo da base de código, vamos enviar objetos Job do Kubernetes ao cluster.
Forneça as variáveis INPUT_FILEPATH e OUTPUT_FILEPATH ao shell script, e ele vai lançar um job FastQC usando um dos arquivos FASTQ que você obteve antes, salvando, no fim, o relatório compactado em um bucket. Como alternativa, você pode substituir as variáveis no arquivo fastqc.yaml manualmente e rodar o job com: kubectl create -f fastqc.yaml
Acompanhe o progresso do FastQC com: kubectl get pods -n biojobs
Você vai notar que um único pod é lançado, que começa baixando a imagem do container e, logo depois, executa o job especificado:

Pod FastQC — status ContainerCreating

Pod FastQC — status Running
Verifique os logs da aplicação enquanto ela roda com kubectl logs -n biojobs <pod_name>:
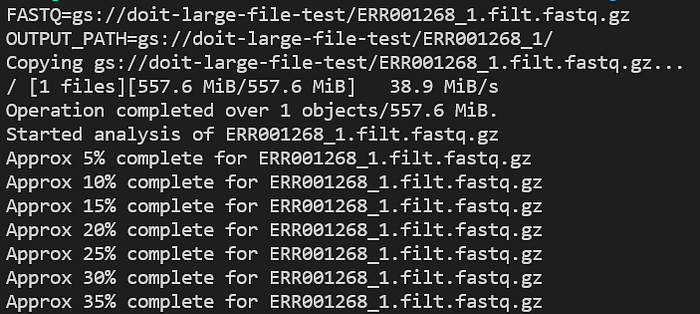
Progresso do pod FastQC
Como os containers — e o GKE/EKS — são construídos pensando em monitoramento, dá para localizar facilmente essas entradas de log na solução de logging nativa da sua nuvem, possibilitando retenção de longo prazo e capacidade investigativa.
Use a frase complete for dentro do Cloud Logging do GCP ou do CloudWatch da AWS para localizar entradas de log do pod FastQC:
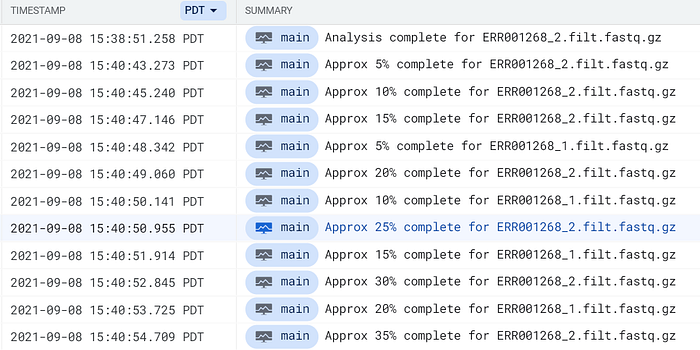
Logs do pod FastQC chegando ao Cloud Logging do GCP
Em algum momento, você verá um relatório FastQC chegar ao caminho do seu bucket de saída. Tente rodar pods FastQC para os dois arquivos FASTQ até ver os relatórios:
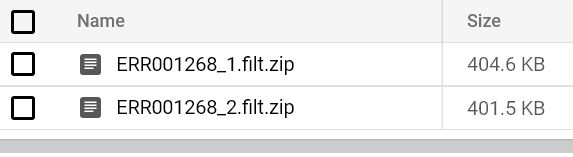
Relatórios FastQC armazenados no bucket
É só isso para rodar FastQC em escala no Kubernetes!
Você pode lançar um job FastQC ou dez mil de uma vez. O plano de controle do Kubernetes, junto com o serviço de auto-escalonamento de nós de computação associado ao seu ambiente de nuvem, vai aumentar e diminuir a capacidade de computação conforme necessário para atender à demanda dos workloads, conforme especificado nos requisitos de cpu/memória/disco definidos no arquivo YAML do Object Kubernetes do FastQC.
Só garanta que os valores máximos permitidos pelo cluster autoscaler para contagem de nós / cpu / memória estejam altos o suficiente — ou pelo menos onde você quer que eles estejam para fins de controle de custos — na criação do cluster. (GCP: gke.tf AWS: cluster.tf)
Você pode rodar bwa-mem2 em escala, com bom custo-benefício, com a mesma facilidade!
Para GCP, vá até GCP/gke/bwa-mem2/
Para AWS, vá até AWS/eks/bwa-mem2/
Forneça os caminhos de entrada/saída para as variáveis no topo de run_bwa-mem2_job.sh e elas vão substituir os nomes de variáveis em bwa-mem2.yaml, em seguida envie o Job ao seu cluster.
Continue monitorando o status dos pods e os logs até ver um arquivo BAM no caminho do seu bucket de saída:
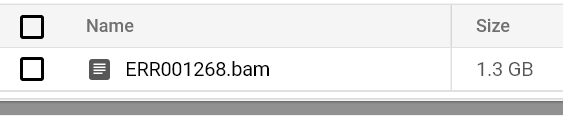
Arquivo BAM armazenado no bucket
Se você verificar as métricas de utilização de CPU dos nós GCE / EC2 que sustentam seu cluster, vai notar que os jobs FastQC, de baixa intensidade e single-core, são enviados a máquinas com CPU menor, enquanto os jobs BWA, que utilizam todos os núcleos, são enviados a máquinas com alta CPU. Eles correspondem aos node groups Kubernetes de baixa/alta CPU criados para garantir que máquinas adequadamente dimensionadas, sustentando cada workload drasticamente diferente, escalem para cima e para baixo de forma eficiente em custos.
Por exemplo, na AWS, não faria sentido provisionar uma máquina m5.24xlarge de 96 vCPUs ao escalar apenas para rodar um novo job FastQC single-core, então enviamos o FastQC para um node group com máquinas m5.xlarge de 4 vCPUs. Já o bwa precisa do máximo de núcleos possível, então enviamos esses jobs para o node group de 96 vCPUs, que escala de forma independente do node group de 4 vCPUs (FastQC).
Para quem já construiu workflows de análise secundária que pegam dados brutos de sequenciamento e os processam para descobrir variantes, o poder do Kubernetes provavelmente está começando a ficar evidente agora. Acabou a era de:
- Job schedulers on-prem com performance instável em escala
- Se preocupar se lançar muitos jobs de uma vez vai derrubar o job scheduler ou até o próprio cluster que sustenta seus workloads (e os dos outros).
- Irritar seus colegas ao tirar capacidade de computação on-prem dos workloads que eles querem rodar.
- Ter que monitorar e relançar workloads que falham por motivos inesperados, mas recuperáveis.
- Arrancar os cabelos tentando descobrir arquivos de log de jobs que travaram — frequentemente em aplicações sem uma funcionalidade de log bem implementada.
Agora, se ao menos você não precisasse escrever código espaguete encadeando as etapas do pipeline…
Passo 5: Envie workflows do Argo (pipelines de jobs Kubernetes)
O Argo permite encadear jobs em pipeline de uma forma que se mantém operacional em escala e suporta novas tentativas tanto de jobs quanto de pipelines em caso de falhas inesperadas.
Vamos demonstrar como usar o argo para rodar dois pods FastQC simultaneamente. Em seguida, se e somente se esses dois pods forem concluídos com sucesso, é lançado o alinhamento com bwa-mem2, usando o mesmo par de FASTQ.
(Como exercício pessoal: dá para considerar adicionar uma etapa de aprovação/reprovação à imagem do FastQC, de modo que, se o FASTQ tiver qualidade tão baixa que não vale a pena fazer o alinhamento, a etapa BWA neste workflow argo seja ignorada.)
Para GCP, vá até GCP/gke/argo_workflow/
Para AWS, vá até AWS/eks/argo_workflow/
Forneça os caminhos de entrada/saída para as variáveis no topo de run_fastqc_to_bwa.sh e elas vão substituir os nomes de variáveis em fastqc_to_bwa.yaml, em seguida envie o Job ao seu cluster.
Execute continuamente kubectl get pods -n biojobs e você verá os pods FastQC rodando, depois concluindo, seguidos pelo lançamento de um job BWA:

Pods FastQC rodando nos arquivos FASTQ R1 e R2 da NA12878
…
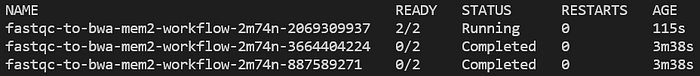
Pod de alinhamento bwa-mem2 lançado depois que os dois pods FastQC concluíram
Vamos olhar o pod bwa-mem2 com:
kubectl logs -n biojobs <pod_name> main
(Note que ‘main’ foi adicionado depois do nome do pod. Com o argo, há logs de pod ‘main’ e ‘wait’ para cada job em um workflow.)
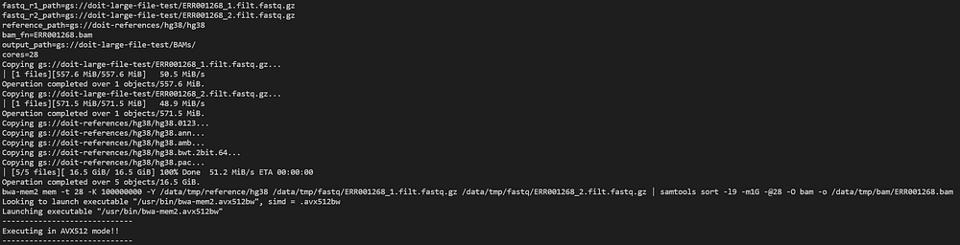
bwa-mem2 trabalhando como parte de um workflow do argo
Você pode rodar kubectl get wf -n biojobs para monitorar o progresso do workflow do argo:

No fim, quando o BWA terminar, o workflow estará concluído, e você vai encontrar um par de relatórios FastQC e um BAM no seu bucket de saída.
Passo 6: Limpeza
Provisionamos uma infraestrutura de computação razoavelmente cara para sustentar o BWA; não queremos esse cluster GKE/EKS rodando à toa quando não está em uso.
Basta percorrer as pastas Terraform na ordem inversa e executar:
terraform destroy -auto-approve
Isso não vai remover os buckets que você criou manualmente para servir como backend do Terraform e para armazenar os arquivos FASTQ e de referência. O Terraform só conhece e consegue monitorar mudanças em recursos que ele mesmo provisionou. No GCP, você também vai precisar excluir manualmente as imagens de container no GCR.
Para GCP, isso significa rodar o comando destroy nesta ordem:
GCP/terraform/standard/kubectl_commands/
GCP/terraform/standard/
Para AWS, isso significa rodar o comando destroy nesta ordem:
AWS/terraform/eks/kubectl_commands/
AWS/terraform/eks/
AWS/terraform/ecr/ (se quiser deletar os repositórios de imagens)
AWS/terraform/
Passo 7: Próximos passos
Com as habilidades aprendidas até aqui, você pode construir pipelines de complexidade arbitrária, baseados em qualquer aplicação, já que qualquer programa pode ser containerizado.
Com código de imagem de container versionado, containers com tags de versão, infraestrutura versionada e pipelines argo, dá para implantar novas versões de software e de pipeline, descontinuar versões antigas e fazer rollback durante uma implantação com defeito, com relativa facilidade — pelo menos em comparação com as soluções alternativas — caso siga este caminho de aprendizado.
Cabe a você decidir quais são os próximos passos, mas espero ter deixado você com as habilidades essenciais para a computação científica moderna e escalável.
As melhores práticas só te levam até certo ponto na nuvem. Construir pensando em alta disponibilidade, resiliência a falhas e escalabilidade otimizada em custo é extremamente desafiador hoje. Essas tarefas adicionais de engenharia se somam à pilha cada vez maior de workloads, algo que afeta diretamente a produtividade.
Como cientista no século 21, é essencial construir aplicações pensando em escalabilidade e facilidade de implantação. Não fazer isso só cria dor de cabeça para a sua organização. No longo prazo, esses problemas viram bola de neve e se transformam em questões sérias e intermináveis em produção.
Se você se inspirou neste projeto, mas também se sente um pouco sobrecarregado, fale com a DoiT International . Você também pode me mandar mensagem no LinkedIn .
A DoiT se orgulha de orientar clientes rumo a arquiteturas de nuvem com custo otimizado, escaláveis, altamente disponíveis, resilientes e com capacidade para big data — um serviço oferecido literalmente sem custo. Recebemos o prêmio de Google Cloud 2020 Global Reseller Partner of the Year por um bom motivo.
Conquistamos a distinção de certificação da Amazon Partner Network (APN), mantendo-nos como AWS Advanced Consulting Partner, graças a um time de engenharia de altíssimo nível e a um forte suporte ao cliente. Todas as informações são comunicadas a você sob demanda, para responder a perguntas relacionadas à nuvem ou resolver problemas em andamento.
Muito obrigado pelo seu tempo. Espero que estes dois artigos tenham ajudado a aprimorar suas habilidades. Crescendo em escala? Entre em contato e deixe a gente te ajudar a alcançar seus objetivos.
Obrigado pela leitura! Para se manter conectado, siga-nos no DoiT Engineering Blog , no canal da DoiT no LinkedIn e no canal da DoiT no Twitter . Para conhecer oportunidades de carreira, acesse https://careers.doit-intl.com .