Benvenuti nella Parte 2 di Scientific Cloud Computing su scala globale con Kubernetes e Terraform. In questo articolo presento una demo completa di scientific cloud computing su scala globale con Kubernetes e Terraform. Per rinfrescare la memoria o approfondire principi e concetti del DevOps moderno, vi rimando alla Parte 1 di questa serie.
Questa guida illustra come DevOps, container, Kubernetes e Terraform possano essere utilizzati nel deployment reale di workloads di calcolo.
PerfectScale™ for Kubernetes
Ready to optimize?
Get your free Kubernetes savings analysis
Panoramica
A grandi linee, il codice di esempio:
- Mostra come confezionare strumenti di bioinformatica diffusi (FastQC e BWA) in container image e caricarli sul repository di immagini di ciascun cloud
- Avvia (e consente di smantellare rapidamente) tutta l'infrastruttura cloud necessaria per eseguire i workloads tramite Terraform, uno strumento di Infrastructure as Code
- Distribuisce una pipeline di workflow basata su queste immagini e sull'infrastruttura cloud appena creata sul servizio Kubernetes completamente gestito di ciascun cloud, un sistema di gestione e orchestrazione dell'esecuzione dei container. Per orchestrare un workflow o una pipeline di task su un cluster Kubernetes utilizzeremo Argo
Combinando Docker, Terraform, Kubernetes e Argo, imparerete a:
- Avviare, aggiornare e dismettere l'infrastruttura necessaria per eseguire i vostri workloads con pochi semplici comandi
- Eseguire workloads end-to-end orchestrati come DAG con retry automatici dei singoli step in caso di errori imprevisti o guasti dell'infrastruttura
- Centralizzare il logging dei workloads e il monitoraggio delle metriche negli strumenti cloud-native nativi per l'esplorazione di log e metriche
- Eseguire workloads su un'infrastruttura che scala automaticamente verso l'alto (fino a capacità su scala globale) e verso il basso (fino a poche o zero risorse di calcolo) in base alle esigenze, pagando solo per le risorse di CPU/RAM/storage effettivamente necessarie a portare a termine i task. Sono finiti i tempi dei server lasciati accesi a vuoto, che facevano lievitare la fattura senza motivo
- Distribuire senza intoppi nuove versioni del software e dismettere progressivamente quelle obsolete
Your cloud bill shouldn't be a mystery
Optimization, automation, expertise. In one platform.
Walkthrough del codice
Acquisite le nozioni di base sul DevOps, vediamo come il codice associato a questo articolo metta in pratica i principi fondamentali del DevOps. Questo walkthrough mostrerà come costruire l'infrastruttura per eseguire pipeline scientifiche capaci di operare su scala globale. Nello specifico, eseguiremo i componenti di una tipica pipeline di bioinformatica.
L'obiettivo è:
- Generare report su una coppia di file di dati grezzi di sequenziamento del genoma per validarne la qualità. I dati grezzi di sequenziamento sono memorizzati in formato FASTQ e i controlli QC vengono eseguiti con uno strumento chiamato FastQC.
- Una volta prodotti i report, avviare un processo noto come allineamento, che in ultima analisi (tramite ulteriori elaborazioni a valle) consente di individuare le mutazioni. Questa fase si esegue con uno strumento chiamato bwa-mem2.
In altre parole, vogliamo che la nostra pipeline basata su Kubernetes esegua simultaneamente due container FastQC su una coppia di file di sequenziamento FASTQ; se entrambi i task vanno a buon fine, deve poi eseguire l'allineamento bwa-mem2 della coppia di dataset di sequenziamento grezzi rispetto alla versione più recente del genoma umano, nota come hg38. Sia i report FastQC sia il file di allineamento prodotto da BWA, noto come file BAM, verranno caricati in un bucket per l'archiviazione a lungo termine.
Attenzione ai costi
Poiché questo codice vi guida attraverso un caso d'uso analitico reale e non un esempio banale, è prevedibile che la configurazione dell'ambiente di calcolo abbia un costo non trascurabile. A seconda di quanto a lungo lascerete in esecuzione l'infrastruttura Terraform, in genere è plausibile attendersi una spesa a due cifre in dollari sulla fattura cloud.
Step 0: Installare i client locali: Cloud CLI, Terraform e kubectl
Assicuratevi che lo strumento Cloud CLI sia installato in locale e autenticato sull'account che intendete utilizzare. Link CLI: GCP e AWS. Avviare l'infrastruttura cloud-as-code è semplice: basta posizionarsi in una cartella di file YAML Terraform ed eseguire:
terraform apply -auto-approve
Perché funzioni, occorre installare il client Terraform in locale. Terraform utilizzerà automaticamente le credenziali di autenticazione della Cloud CLI.
Eseguire un job o un workflow su Kubernetes è altrettanto semplice: basta creare il template YAML del job/workflow ed eseguire:
kubectl create -f job.yaml
Perché funzioni, occorre installare il client kubectl in locale. kubectl viene autenticato con un comando CLI specifico per il cloud.
Step 1: Acquisire i dati FASTQ e il riferimento umano (hg38)
Prima di poter mettere in pratica questo workflow, vi serviranno una coppia di file FASTQ di esempio e il genoma di riferimento umano hg38.
Per contenere al minimo i costi di elaborazione lavorando comunque con dati su scala reale, ho prelevato due file FASTQ da circa 0,5 GB ciascuno relativi al campione 'gold standard' NA12878 dal bucket S3 del progetto 1000 Genomes. Consiglio vivamente di creare un bucket GCS o S3 nel vostro account cloud e di copiarvi questi file. Se testate il workflow più volte, è meglio evitare di scaricare ripetutamente dal bucket S3 di 1000G.
Tenete sempre presenti i costi di trasferimento dati cross-region .
I file FASTQ si trovano nella regione us-east-1 di Amazon, quindi spostare questi dati verso un altro bucket S3 regionale (ad esempio la regione us-west-2 utilizzata nei miei test) o fuori dal cloud AWS verso un bucket Cloud Storage di GCP comporta un costo per ogni GB di dati trasferito al di fuori della regione del bucket. Create un bucket nella regione cloud desiderata e copiate al suo interno i FASTQ:
# R1 FASTQ in un bucket S3 us-east-1
s3://1000genomes/phase3/data/NA12878/sequence_read/ERR001268_1.filt.fastq.gz
# R2 FASTQ in un bucket S3 us-east-1
s3://1000genomes/phase3/data/NA12878/sequence_read/ERR001268_2.filt.fastq.gz# Esempio di comando per copiare un oggetto:
YOUR_FASTQ_BUCKET=<BUCKET_NAME_HERE># Per AWS, usa:
aws s3 cp s3://1000genomes/phase3/data/NA12878/sequence_read/ERR001268_1.filt.fastq.gz s3://{YOUR_FASTQ_BUCKET}/FASTQ/
# Per GCP, copia il file nello storage locale di una VM con 'aws s3 cp'
# Poi copia dallo storage locale a un bucket GCS con gsutil:
gsutil -o GSUtil:parallel_composite_upload_threshold=150M cp /tmp/FASTQ/ERR001268_1.filt.fastq.gz gs://{YOUR_FASTQ_BUCKET}/FASTQ/
Dovrete anche scaricare l'ultima versione del genoma di riferimento umano e costruirne un indice bwa-mem2. Consiglio di:
- Copiare il FASTA hg38 dal bucket S3 us-east-1 del Broad Institute su una VM con elevata memoria nel cloud che preferite. Può essere una macchina r5.16xlarge su AWS o una n2-highmem-64 su GCP.
- Installare bwa-mem2 (oppure utilizzare la container image descritta più avanti)
- Costruire l'indice di riferimento bwa-mem2 (richiede 1–2 ore)
- Caricare i file di output in un nuovo bucket sul vostro account cloud
# genoma di riferimento hg38 dal Broad Institute, in un bucket S3 us-east-1
s3://broad-references/hg38/v0/Homo_sapiens_assembly38.fasta# Esempio di comando per costruire l'indice bwa-mem2 e salvarlo nel bucket
YOUR_REF_BUCKET=<BUCKET_NAME_HERE># Sia per AWS che per GCP, usa:
aws s3 cp s3://broad-references/hg38/v0/Homo_sapiens_assembly38.fasta /tmp/reference/hg38.fastabwa-mem2 index -p hg38 /tmp/reference/hg38.fasta# Per AWS, usa:
aws s3 sync /tmp/reference/ s3://${YOUR_REF_BUCKET}/hg38/
# Per GCP, usa:
gsutil -m -o GSUtil:parallel_composite_upload_threshold=150M cp "/tmp/reference/hg38*" gs://${YOUR_REF_BUCKET}/hg38/
Se avete seguito correttamente i passaggi precedenti, ora avete due FASTQ (dati grezzi di sequenziamento del genoma) e il genoma di riferimento umano hg38 pronti per i test del workflow di bioinformatica.
Step 2: Costruire l'infrastruttura cloud
Con Terraform avvieremo tutta l'infrastruttura cloud necessaria a eseguire un cluster Kubernetes completamente gestito ed eseguiremo alcuni comandi sul cluster (tramite kubectl) per installare argo, uno strumento Kubernetes per l'esecuzione di workflow (ad esempio pipeline di bioinformatica).
Per impostazione predefinita, Terraform mantiene lo stato dell'infrastruttura cloud in locale; tuttavia, in un caso d'uso reale conviene utilizzare un bucket come backend Terraform: in questo modo, quando le persone in azienda apportano una modifica all'infrastruttura, tutti operano sullo stesso dataset di 'verità' aggiornato che descrive lo stato dell'infrastruttura.
L'utilizzo di un bucket come backend blocca inoltre le modifiche all'infrastruttura mentre un altro set di modifiche è in fase di applicazione, evitando così l'esecuzione simultanea di più operazioni Terraform potenzialmente in conflitto. Per abilitare un backend Terraform basato su bucket, create un bucket nel vostro ambiente cloud, quindi sostituite tutti i riferimenti a doit-matt-tf-state-test nel codice con il nome del vostro bucket.
Con il bucket di backend Terraform pronto, potete avviare l'infrastruttura.
Se intendete eseguire il vostro workload su Google Cloud, posizionatevi in GCP/terraform. All'interno di questa cartella ci sono le sottocartelle standard e autopilot, ciascuna contenente codice TF che lancerà un cluster GKE in modalità GKE Standard o Autopilot. Autopilot è in genere l'opzione preferita, perché non solo orienta i nodi (risorse di calcolo) verso un modello serverless, ma applica anche numerose best practice di sicurezza.
Purtroppo, Autopilot non funziona in questa demo a causa di una limitazione: lo spazio disco effimero è limitato a 10 GiB, quantità non sufficiente per eseguire FastQC né bwa-mem2 su dati reali. Tuttavia, qualora questa limitazione venisse rimossa in futuro, l'utilizzo di Autopilot rispetto a Standard è preferibile. Ho incluso il codice TF per Autopilot proprio nel caso in cui questa limitazione venga aumentata dopo la pubblicazione. Anche AWS ECS Fargate ha una limitazione di spazio disco di 200 GiB, non sufficiente per la maggior parte degli usi reali di scientific computing. Il codice TF per Fargate non è incluso nel repository.
Se intendete eseguire il vostro workload su AWS, posizionatevi in AWS/terraform.
Una volta nella cartella terraform specifica per il vostro cloud, eseguite quanto segue:
terraform apply -auto-approve# Esegui prima 'terraform plan' per visualizzare l'elenco delle modifiche infrastrutturali pianificate senza creare nulla
Questo pianificherà ed eseguirà immediatamente l'insieme dei file '.tf' di Terraform presenti nella cartella per avviare un cluster K8s e le relative risorse prerequisite. Terraform chiederà un GCP Project ID o un AWS Account ID, e nient'altro.
Potete eventualmente modificare il file variables.tf prima di eseguire apply, ad esempio per cambiare la regione in cui verrà lanciato il cluster, il nome del cluster o per impostare in modo fisso la variabile project GCP/account ID AWS.
Vi consiglio vivamente di esaminare il codice per imparare Terraform
Per accelerare la vostra comprensione, su GCP vengono creati i seguenti elementi:
- Abilitazione delle API Compute, Container e IAM
- Un cluster GKE Standard con auto-scaling all'interno della VPC predefinita, che utilizza la versione di Kubernetes predefinita e aggiornata (v1.20 al momento della pubblicazione) con alcune funzionalità di sicurezza opzionali abilitate, come Workload Identity, Shielded Nodes e Secure Boot. È un cluster regionale con nodi distribuiti su più zone (data center)
- Un Service Account GCP (SA) e il relativo Kubernetes Service Account (KSA) collegato che abilitano Workload Identity per limitare lo scope dei permessi GCP per tutti i job eseguiti all'interno di un determinato namespace Kubernetes. Il SA/KSA potrà leggere/scrivere sui bucket e scrivere log e metriche su Cloud Logging e Cloud Metrics
- Il cluster utilizza istanze preemptible / Spot. Pagherete un prezzo significativamente inferiore rispetto a quello on-demand, con la possibilità che i nodi vengano terminati inaspettatamente e quindi sostituiti. K8s identificherà automaticamente i job falliti a causa della terminazione improvvisa dei nodi e li ripianificherà
Per AWS vengono creati i seguenti componenti di rete e sicurezza:
- Una nuova VPC dedicata a ospitare il cluster K8s
- Subnet pubbliche (non utilizzate) e private + tabelle di routing per i nodi del cluster
- Un NAT Gateway, endpoint VPC S3 ed ECR con i relativi security group abilitati al TLS, oltre all'abilitazione del supporto DNS hostname per consentire un cluster di nodi privati
- Un Internet Gateway per abilitare l'accesso pubblico al control plane del cluster
Sempre per AWS, dovrete poi posizionarvi in AWS/terraform/ecr ed eseguire nuovamente terraform apply, poiché i file .tf di questa cartella creano:
- I repository in cui verranno memorizzate le container image che andrete a creare
(Su GCP non è necessario creare i repository)
Sempre per AWS, dovrete inoltre posizionarvi in AWS/terraform/eks ed eseguire nuovamente terraform apply, poiché i file .tf di questa cartella creano:
- Un ruolo IAM per il cluster che abilita l'accesso in lettura e scrittura ai bucket S3, oltre all'accesso SSH browser-based e senza chiavi sui nodi tramite SSM Manager
- Un cluster EKS con auto-scaling che utilizza la versione di Kubernetes predefinita e aggiornata (v1.21 al momento della pubblicazione). Workload Identity, Shielded Nodes e Secure Boot non sono funzionalità immediatamente disponibili su EKS. È un cluster regionale con nodi distribuiti su più zone (data center)
- Il cluster utilizza istanze preemptible / Spot. Pagherete un prezzo significativamente inferiore rispetto a quello on-demand, con la possibilità che i nodi vengano terminati inaspettatamente e quindi sostituiti. K8s identificherà automaticamente i job falliti a causa della terminazione improvvisa dei nodi e li ripianificherà
Una volta completate le operazioni di terraform apply, il cluster è operativo e pronto a ricevere comandi API Kubernetes tramite kubectl. Basta autenticare kubectl con un comando specifico per il cloud:
# Per GCP:
gcloud container clusters get-credentials --region=us-central1 bioinformatics-tasks
# Per AWS:
aws eks update-kubeconfig --name=bioinformatics-tasks
A questo punto potrete eseguire vari comandi sul cluster, ad esempio elencare i pod distribuiti in tutti i namespace con kubectl get pods -A:
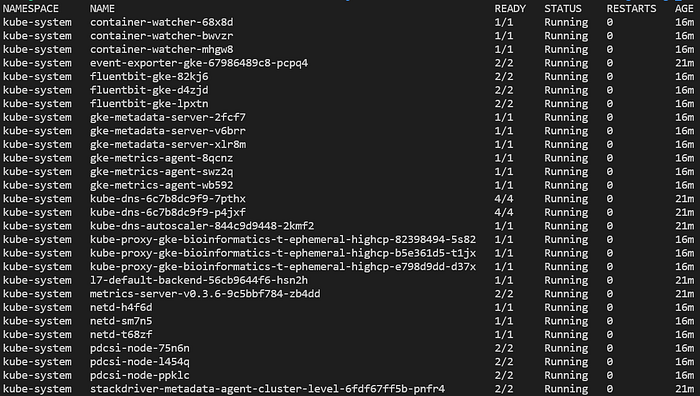
Pod tipici in un nuovo cluster GKE Standard completamente gestito
Avviato il cluster, dobbiamo ora installarvi argo, perché ci permetterà di inviare non solo singoli task, ma anche un workflow / pipeline di task organizzato come DAG.
Per farlo su GCP, posizionatevi in GCP/terraform/standard/kubectl_commands
Per farlo su AWS, posizionatevi in AWS/terraform/kubectl_commands
L'esecuzione di terraform apply -auto-approve in queste cartelle eseguirà comandi kubectl gestiti da Terraform che:
- Creano un namespace 'biojobs' a cui verranno inviati i job, con uno scope di permessi (su GCP) limitato alla sola lettura/scrittura di oggetti da un bucket.
- Installano argo e, su GCP con la sua Workload Identity, gli concedono permessi adeguatamente delimitati per monitorare l'avanzamento dei pod nel namespace 'biojobs'.
Potete verificare che il namespace sia stato creato e argo installato elencando tutti i namespace e poi interrogando i pod in esecuzione nel namespace argo:
kubectl get ns
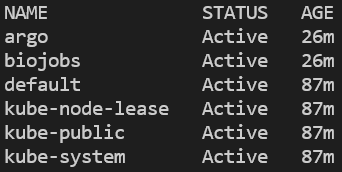
Tutti i namespace del cluster. Su EKS comparirà anche 'amazon-cloudwatch.'
kubectl get pods -n argo
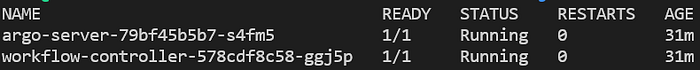
Pod argo server e workflow controller in esecuzione nel namespace 'argo'
Infine, eseguite questo comando per assicurarvi che argo sia compatibile con il passaggio di Kubernetes ≥v1.19 dal container runtime docker a containerd. Non preoccupatevi di questo dettaglio: argo risolverà il problema in futuro:
kubectl patch configmap workflow-controller-configmap -n argo --patch ‘{"data":{"containerRuntimeExecutor":"k8sapi"}}’
Con argo installato e i nodi di calcolo pronti, diamo libero sfogo alla creatività!
Step 3: Costruire le container image di FastQC e bwa-mem2
Per eseguire i workloads di FastQC e bwa-mem2 su AWS o GCP, dobbiamo costruire le container image di questi strumenti e memorizzarle nel repository di immagini container del rispettivo cloud (GCR per GCP, ECR per AWS).
Posizionatevi in images/fastqc/
All'interno di questa cartella si trova il Dockerfile che definisce come confezionare FastQC in un container insieme a s5cmd (uno strumento per copie veloci AWS S3 ←→ locale) e gsutil (per copie GCS ←→ locale). Dedicate del tempo a esaminare il contenuto del file e ad approfondire come si costruiscono i Dockerfile. A margine, potreste essere interessati a questo articolo che ho scritto sulla massimizzazione della velocità di trasferimento dati dei bucket: i principi descritti sono utilizzati nel Dockerfile.
Eseguite build_fastqc_image_gcp.sh oppure build_fastqc_image_aws.sh per creare una container image dal Dockerfile e farne push, rispettivamente, su GCR o ECR.
Per creare una container image per bwa-mem e memorizzarla in GCR o ECR, posizionatevi in images/bwa-mem2/ e ripetete lo stesso processo di esecuzione dello script di build.
Completato questo step, FastQC e bwa-mem2 sono pronti per essere eseguiti su un cluster Kubernetes tramite le container image memorizzate nel repository di immagini del vostro ambiente cloud.
Step 4: Inviare e monitorare i job Kubernetes
Passiamo ora alla cartella specifica del vostro cloud per i job Kubernetes e ne lanciamo alcuni. In seguito eseguiremo FastQC e bwa-mem2 in pipeline con argo, ma per cominciare li eseguiremo come task separati.
Per GCP, posizionatevi in GCP/gke/FastQC/
Per AWS, posizionatevi in AWS/eks/FastQC/
All'interno di queste cartelle si trova un Job Kubernetes denominato fastqc.yaml.
Questo YAML definisce un job eseguibile con kubectl create -f fastqc.yaml. Tuttavia, il file contiene alcune variabili che devono essere definite e sostituite da run_fastqc_job.sh. Dedicate del tempo a comprendere gli objects Kubernetes in generale e i Job nello specifico per decifrare fastqc.yaml, e analizzate inoltre cosa avviene nello shell script.
In sostanza, Kubernetes viene usato più spesso per eseguire pod a lunga durata chiamati Deployment (ad esempio web server long-lived), ma può anche eseguire Job, ovvero task con esecuzione una tantum, come programmi scientifici quali FastQC. In tutto il codice invieremo oggetti Job Kubernetes al cluster.
Fornite le variabili INPUT_FILEPATH e OUTPUT_FILEPATH allo shell script, che lancerà un job FastQC utilizzando uno dei file FASTQ scaricati in precedenza, salvando infine il report compresso in un bucket. In alternativa, potete sostituire manualmente le variabili nel file fastqc.yaml ed eseguire il job con: kubectl create -f fastqc.yaml
Tenete sotto controllo l'avanzamento di FastQC con: kubectl get pods -n biojobs
Noterete che viene lanciato un singolo pod, che inizia scaricando la container image e subito dopo esegue il job specificato:

Pod FastQC — stato ContainerCreating

Pod FastQC — stato Running
Consultate i log dell'applicazione mentre è in esecuzione con kubectl logs -n biojobs <pod_name>:
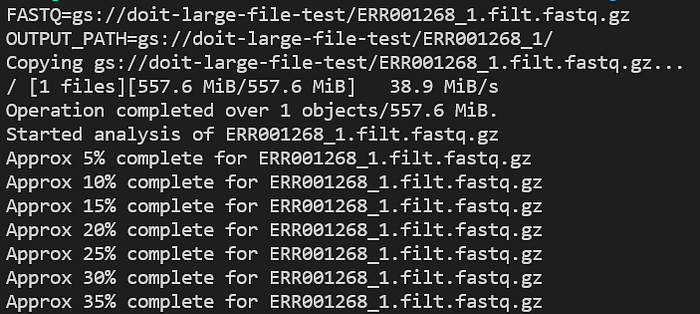
Avanzamento del pod FastQC
Poiché i container — e GKE/EKS — sono progettati con il monitoring in mente, potete individuare facilmente queste voci di log nella soluzione di logging nativa del vostro cloud, abilitando la conservazione dei log a lungo termine e la possibilità di indagini approfondite.
Cercate la stringa complete for in Cloud Logging di GCP o in CloudWatch di AWS per trovare le voci di log dei pod FastQC:
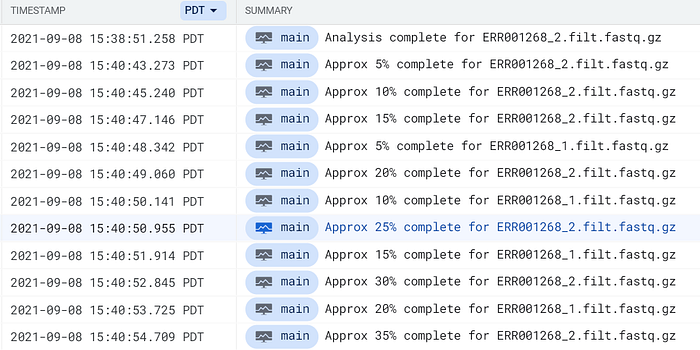
Log dei pod FastQC che arrivano in Cloud Logging di GCP
Prima o poi vedrete arrivare un report FastQC nel percorso del bucket di output. Provate a eseguire i pod FastQC su entrambi i file FASTQ finché non vedete i rispettivi report:
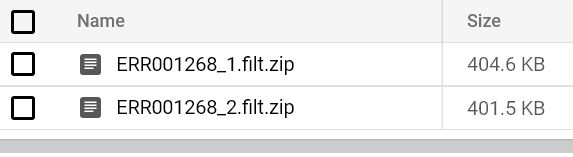
Report FastQC archiviati nel bucket
Ed è tutto ciò che serve per eseguire FastQC su scala con Kubernetes!
Potete lanciare un solo job FastQC oppure diecimila contemporaneamente. Il control plane di Kubernetes, insieme al servizio di auto-scaling dei nodi di calcolo del relativo ambiente cloud, aumenterà e ridurrà la capacità di calcolo in base alle esigenze dei workloads, secondo i requisiti di cpu/memoria/disco definiti nel file YAML del Kubernetes Object di FastQC.
Assicuratevi soltanto che i valori massimi consentiti dall'autoscaler del cluster per numero di nodi / cpu / memoria siano sufficientemente alti — o almeno al livello desiderato per il controllo dei costi — al momento della creazione del cluster. (GCP: gke.tf AWS: cluster.tf)
Anche bwa-mem2 può essere eseguito in modo cost-effective e su scala con la stessa facilità!
Per GCP, posizionatevi in GCP/gke/bwa-mem2/
Per AWS, posizionatevi in AWS/eks/bwa-mem2/
Indicate i percorsi di input/output per le variabili in cima a run_bwa-mem2_job.sh che sostituiranno i nomi delle variabili in bwa-mem2.yaml, quindi inviate il Job al cluster.
Continuate a monitorare lo stato dei pod e i log finché non vedrete un file BAM in output nel percorso del vostro bucket di output:
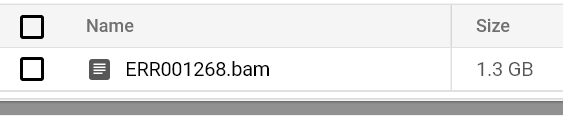
File BAM archiviato nel bucket
Se controllate le metriche di utilizzo della CPU dei nodi GCE / EC2 che alimentano il cluster, noterete che i job FastQC, a bassa intensità e single-core, vengono inviati a macchine con CPU più piccole, mentre i job BWA, che sfruttano tutti i core, vengono inviati a macchine con CPU elevata. Si tratta dei gruppi di nodi Kubernetes a CPU bassa/alta creati per garantire che ogni workload, di natura profondamente diversa, sia alimentato da macchine di dimensioni adeguate, scalando verso l'alto e verso il basso in modo cost-effective.
Ad esempio, su AWS non avrebbe senso provisionare una macchina m5.24xlarge da 96 vCPU durante lo scale-up solo per eseguire un nuovo job FastQC single-core, quindi inviamo FastQC a un gruppo di nodi composto da macchine m5.xlarge da 4 vCPU. bwa, invece, richiede tutti i core che riuscite a fornirgli, perciò inviamo questi job al gruppo di nodi da 96 vCPU, che scalerà in modo indipendente rispetto al gruppo da 4 vCPU dedicato a FastQC.
Chi di voi ha già costruito workflow di analisi secondaria che prendono dati grezzi di sequenziamento e li elaborano per individuare varianti probabilmente comincerà ora a intuire la potenza di Kubernetes. Sono finiti i tempi di:
- Job scheduler on-prem dalle prestazioni capricciose su scala
- Preoccuparsi che il lancio di troppi job in contemporanea possa mettere in ginocchio il job scheduler, o addirittura il cluster stesso, che alimenta i vostri workloads (e quelli di tutti gli altri).
- Far innervosire i colleghi sottraendo capacità di calcolo on-prem ai workloads che vorrebbero eseguire.
- Dover monitorare e rilanciare workloads che falliscono per motivi imprevisti ma recuperabili.
- Strapparsi i capelli per recuperare i file di log dai job andati in crash — spesso per applicazioni con funzionalità di logging implementate male.
Adesso, se solo non si dovesse scrivere codice spaghetti per concatenare gli step in pipeline…
Step 5: Inviare workflow Argo (pipeline di job Kubernetes)
Argo permette di concatenare i job in pipeline in modo da restare operativi su scala e di gestire il retry sia dei singoli job sia dell'intera pipeline in caso di guasti imprevisti.
Vediamo come usare argo per eseguire due pod FastQC in contemporanea. Poi, soltanto se entrambi i pod si completano con successo, viene lanciato l'allineamento con bwa-mem2 sulla stessa coppia di FASTQ.
(Come esercizio personale: potreste pensare di aggiungere uno step pass/no-pass alla container image di FastQC, in modo che, se il FASTQ è di qualità troppo bassa per giustificare l'allineamento, lo step BWA del workflow argo venga saltato.)
Per GCP, posizionatevi in GCP/gke/argo_workflow/
Per AWS, posizionatevi in AWS/eks/argo_workflow/
Indicate i percorsi di input/output per le variabili in cima a run_fastqc_to_bwa.sh, che sostituiranno i nomi delle variabili in fastqc_to_bwa.yaml, quindi inviate il Job al cluster.
Eseguite ripetutamente kubectl get pods -n biojobs e vedrete i pod FastQC in esecuzione, poi completarsi e infine lasciare spazio al lancio di un job BWA:

Pod FastQC in esecuzione sui file FASTQ R1 e R2 di NA12878
…
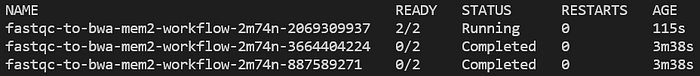
Pod di allineamento bwa-mem2 avviato dopo il completamento dei due pod FastQC
Diamo un'occhiata al pod bwa-mem2 con:
kubectl logs -n biojobs <pod_name> main
(Notate che 'main' è stato aggiunto dopo il nome del pod. Con argo, per ogni job di un workflow esistono i log dei pod 'main' e 'wait'.)
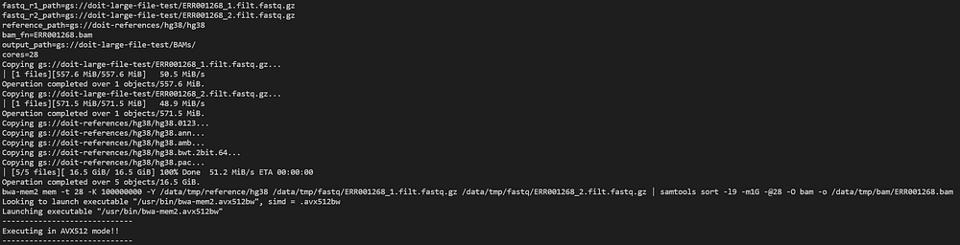
bwa-mem2 al lavoro all'interno di un workflow argo
Potete eseguire kubectl get wf -n biojobs per monitorare l'avanzamento del workflow argo:

Al termine di BWA, anche il workflow risulterà completato, e troverete una coppia di report FastQC e un BAM nel vostro bucket di output.
Step 6: Pulizia
Abbiamo avviato un'infrastruttura di calcolo piuttosto costosa per alimentare BWA; non vogliamo che questo cluster GKE/EKS resti attivo quando non è in uso.
Basta percorrere in ordine inverso le cartelle Terraform ed eseguire:
terraform destroy -auto-approve
Tenete presente che questo non rimuoverà i bucket creati manualmente come backend di Terraform e per archiviare i file FASTQ e di riferimento. Terraform è consapevole soltanto delle risorse che ha avviato e in grado di monitorarne le modifiche. Su GCP dovrete inoltre eliminare manualmente le container image in GCR.
Su GCP, eseguite il comando destroy in questo ordine:
GCP/terraform/standard/kubectl_commands/
GCP/terraform/standard/
Su AWS, eseguite il comando destroy in questo ordine:
AWS/terraform/eks/kubectl_commands/
AWS/terraform/eks/
AWS/terraform/ecr/ (se volete eliminare i repository di immagini)
AWS/terraform/
Step 7: Prossimi passi
Con le competenze acquisite fin qui, potete costruire pipeline di complessità arbitraria basate su qualsiasi applicazione, perché qualunque programma può essere containerizzato.
Con codice di container image versionato, container con tag di versione, infrastruttura versionata e pipeline argo, potete distribuire nuove versioni di software e pipeline, deprecare le versioni precedenti ed eseguire il rollback in caso di deployment difettoso con relativa facilità — perlomeno se confrontato con le soluzioni alternative — qualora decidiate di proseguire su questo percorso di apprendimento.
Sta a voi decidere quali siano i prossimi passi, ma spero di avervi fornito le competenze fondamentali per uno scientific computing moderno e scalabile.
Nel cloud, le best practice possono portarvi solo fino a un certo punto. Progettare per alta disponibilità, resilienza ai guasti e scalabilità ottimizzata sui costi è oggi estremamente impegnativo. Queste attività ingegneristiche aggiuntive si sommano alla mole sempre crescente di workloads, con un impatto diretto sulla produttività.
Per uno scienziato del XXI secolo è essenziale progettare le applicazioni avendo in mente scalabilità e facilità di deployment fin dall'inizio. Non farlo significa creare grattacapi alla propria organizzazione. Sul lungo periodo, questi problemi finiscono per trasformarsi in criticità di produzione gravi e senza fine.
Se questo progetto vi ispira ma allo stesso tempo vi sentite un po' sopraffatti, contattate DoiT International . Potete anche scrivermi su LinkedIn .
DoiT è orgogliosa di guidare i clienti verso architetture cloud cost-optimized, scalabili, ad alta disponibilità, resilienti e capaci di gestire scale di big data, un servizio offerto letteralmente a titolo gratuito. Non a caso siamo stati premiati come Google Cloud 2020 Global Reseller Partner of the Year.
Abbiamo ottenuto la Partner Network (APN) Certification Distinction di Amazon, rimanendo al contempo AWS Advanced Consulting Partner, grazie a Engineers di altissimo livello e a un solido supporto clienti. Le informazioni vengono comunicate on-demand per rispondere a domande sul cloud o risolvere problematiche in corso.
Grazie mille per il vostro tempo. Spero che questi due articoli abbiano contribuito a far crescere le vostre competenze. State scalando? Contattateci e lasciate che vi aiutiamo a raggiungere i vostri obiettivi.
Grazie per la lettura! Per restare in contatto, seguiteci sul DoiT Engineering Blog , sul canale LinkedIn di DoiT e sul canale Twitter di DoiT . Per esplorare le opportunità di carriera, visitate https://careers.doit-intl.com .