Bienvenido a la Parte 2 de Computación científica en la nube a escala global con Kubernetes y Terraform. Aquí te comparto una demo completa de computación científica en la nube a escala global con Kubernetes y Terraform. Si quieres refrescar la memoria o leer más sobre los principios y conceptos modernos de DevOps, pásate a la Parte 1 de esta serie.
Esta guía te mostrará cómo aprovechar DevOps, contenedores, Kubernetes y Terraform en el despliegue real de workloads de cómputo.
PerfectScale™ for Kubernetes
Ready to optimize?
Get your free Kubernetes savings analysis
Panorama general
A grandes rasgos, el código de ejemplo:
- Muestra cómo empaquetar herramientas comunes de bioinformática (FastQC y BWA) en imágenes de contenedor y subirlas al repositorio de imágenes de cada nube
- Levanta (y permite desmontar rápidamente) toda la infraestructura cloud necesaria para ejecutar workloads mediante Terraform, una herramienta de Infraestructura como Código
- Despliega un pipeline de workflow común basado en estas imágenes y la infraestructura cloud levantada en el servicio totalmente gestionado de Kubernetes de cada nube, un sistema de gestión y orquestación de ejecución de contenedores. Se usará Argo para orquestar un workflow o pipeline de tareas en un cluster de Kubernetes
Combinando Docker, Terraform, Kubernetes y Argo, vas a aprender a:
- Levantar, actualizar y desmontar la infraestructura necesaria para correr tus workloads con comandos sencillos
- Ejecutar workloads analíticos dirigidos por DAG de extremo a extremo, con reintentos automáticos de pasos individuales cuando se topan con errores inesperados o fallos de infraestructura
- Centralizar el logging y el monitoreo de métricas de tus workloads en herramientas nativas de la nube para explorar logs y métricas
- Ejecutar workloads sobre infraestructura que escala automáticamente hacia arriba (hasta capacidad a escala global) y hacia abajo (con pocos o ningún recurso de cómputo) según se necesite, de modo que solo pagues por los recursos de CPU/RAM/almacenamiento requeridos para completar tus tareas. Atrás quedaron los días de dejar servidores ociosos inflando tu factura sin necesidad
- Desplegar nuevas versiones de software y retirar las versiones antiguas sin sobresaltos
Your cloud bill shouldn't be a mystery
Optimization, automation, expertise. In one platform.
Recorrido por el código
Con una base de DevOps ya cubierta, veamos cómo el código que acompaña a este post demuestra los principios fundamentales de DevOps. En este recorrido vas a ver cómo construir la infraestructura para soportar la ejecución de pipelines científicos a escala global. En concreto, ejecutaremos componentes de un pipeline común de bioinformática.
Lo que buscamos es:
- Generar reportes sobre un par de archivos de datos crudos de secuenciación genómica que ayuden a validar su calidad. Los datos crudos de secuenciación se almacenan en formato FASTQ, y los chequeos de QC se hacen con una herramienta llamada FastQC.
- Una vez generados esos reportes, realizar un proceso conocido como alineamiento, que en última instancia (mediante el procesamiento posterior con herramientas adicionales) permitirá descubrir mutaciones. Esto se hace con una herramienta llamada bwa-mem2.
Esto significa que queremos que nuestro pipeline basado en Kubernetes ejecute dos contenedores de FastQC sobre un par de archivos de datos de secuenciación FASTQ de manera simultánea y, si ambas tareas terminan con éxito, ejecute el alineamiento bwa-mem2 del par de datasets crudos de secuenciación contra la última versión del genoma humano, conocida como hg38. Tanto los reportes de FastQC como el archivo de alineamiento generado por BWA, conocido como archivo BAM, se subirán a un bucket para almacenamiento a largo plazo.
Aviso sobre el costo
Como este código te lleva a través de un caso analítico real y no de un ejemplo simple, hay que esperar que el entorno de cómputo que se levanta tenga un costo nada despreciable. Dependiendo de cuánto tiempo dejes corriendo la infraestructura de Terraform, en general estimo un gasto de dos dígitos en dólares en tu factura cloud.
Paso 0: Instala los clientes locales: Cloud CLI, Terraform y kubectl
Asegúrate de tener tu CLI de la nube instalada localmente y autenticada contra la cuenta que vas a usar. Enlaces de CLI: GCP y AWS. Levantar infraestructura cloud como código es tan simple como ir a una carpeta con archivos YAML de Terraform y ejecutar:
terraform apply -auto-approve
Para que eso funcione, tendrás que instalar el cliente de terraform localmente. Terraform usará automáticamente las credenciales de autenticación de tu CLI de la nube.
Ejecutar un job o un workflow en Kubernetes es tan sencillo como crear la plantilla YAML para ese job/workflow y correr:
kubectl create -f job.yaml
Para que eso funcione, tendrás que instalar el cliente kubectl localmente. Kubectl se autentica con un comando específico de cada nube.
Paso 1: Conseguir los datos FASTQ y la referencia humana (hg38)
Antes de poder mostrar este workflow, necesitarás un par de archivos FASTQ de ejemplo y el genoma humano de referencia hg38 con el cual trabajar.
Para mantener al mínimo los costos de procesamiento sin renunciar a datos a escala de un caso real, descargué dos archivos FASTQ de ~0.5 GB de la muestra ‘gold standard’ NA12878 desde el bucket S3 del proyecto 1000 Genomes. Recomiendo crear un bucket de GCS o S3 en tu cuenta cloud y copiar estos archivos allí. Mientras pruebas el workflow una y otra vez, seguramente no quieras descargarlos repetidamente desde el bucket S3 de 1000G.
Siempre conviene tener presentes los precios de transferencia de datos entre regiones .
Los archivos FASTQ están en la región us-east-1 de Amazon, así que mover estos datos a otro bucket S3 regional (por ejemplo, la región us-west-2 que usé en mis pruebas) o sacarlos de la nube de AWS hacia un bucket de Cloud Storage de GCP tendrá un costo por cada GB transferido fuera de la región del bucket. Crea un bucket en la región cloud que prefieras y copia los FASTQ allí:
# R1 FASTQ ubicado en un bucket S3 us-east-1
s3://1000genomes/phase3/data/NA12878/sequence_read/ERR001268_1.filt.fastq.gz
# R2 FASTQ ubicado en un bucket S3 us-east-1
s3://1000genomes/phase3/data/NA12878/sequence_read/ERR001268_2.filt.fastq.gz# Comando de ejemplo para copiar un objeto:
YOUR_FASTQ_BUCKET=<BUCKET_NAME_HERE># Para AWS, usa:
aws s3 cp s3://1000genomes/phase3/data/NA12878/sequence_read/ERR001268_1.filt.fastq.gz s3://{YOUR_FASTQ_BUCKET}/FASTQ/
# Para GCP, copia el archivo al almacenamiento local de una VM con 'aws s3 cp'
# Luego, copia desde el almacenamiento local a un bucket de GCS con gsutil:
gsutil -o GSUtil:parallel_composite_upload_threshold=150M cp /tmp/FASTQ/ERR001268_1.filt.fastq.gz gs://{YOUR_FASTQ_BUCKET}/FASTQ/
También necesitarás obtener la última versión del genoma humano de referencia y construir un índice de bwa-mem2 para él. Te recomiendo:
- Copiar el FASTA de hg38 desde el bucket S3 us-east-1 del Broad Institute a una VM de muy alta memoria en la nube de tu elección. Puede ser una máquina r5.16xlarge en AWS o una n2-highmem-64 en GCP.
- Instalar bwa-mem2 (o usar la imagen de contenedor que se describe más adelante)
- Construir el índice de referencia de bwa-mem2 (toma de 1 a 2 horas)
- Subir los archivos de salida a un nuevo bucket en tu cuenta cloud
# Genoma humano de referencia hg38 del Broad Institute ubicado en un bucket S3 us-east-1
s3://broad-references/hg38/v0/Homo_sapiens_assembly38.fasta# Comando de ejemplo para construir el índice de bwa-mem2 y guardarlo en tu bucket
YOUR_REF_BUCKET=<BUCKET_NAME_HERE># Tanto para AWS como para GCP, usa:
aws s3 cp s3://broad-references/hg38/v0/Homo_sapiens_assembly38.fasta /tmp/reference/hg38.fastabwa-mem2 index -p hg38 /tmp/reference/hg38.fasta# Para AWS, usa:
aws s3 sync /tmp/reference/ s3://${YOUR_REF_BUCKET}/hg38/
# Para GCP, usa:
gsutil -m -o GSUtil:parallel_composite_upload_threshold=150M cp "/tmp/reference/hg38*" gs://${YOUR_REF_BUCKET}/hg38/
Si seguiste correctamente los pasos anteriores, ya tendrás dos FASTQ (datos crudos de secuenciación genómica) y el genoma humano de referencia hg38 listos para usarse en las pruebas del workflow bioinformático.
Paso 2: Construye tu infraestructura cloud
Con Terraform levantaremos toda la infraestructura cloud necesaria para correr un cluster de Kubernetes totalmente gestionado, además de ejecutar algunos comandos sobre el cluster (mediante kubectl) que instalan argo en él, una herramienta de Kubernetes para correr workflows (por ejemplo, pipelines de bioinformática).
Por defecto, Terraform mantiene su estado conocido de la infraestructura cloud localmente, pero en un caso real se utiliza un bucket como backend de Terraform. Así, cuando alguien dentro de la empresa hace un cambio en la infraestructura, todos operan desde el mismo dataset actualizado de ‘verdad’ sobre el estado de esa infraestructura.
Usar un bucket como backend también bloquea los cambios mientras se está aplicando otro conjunto de modificaciones, lo que evita que se ejecuten al mismo tiempo varias operaciones de Terraform potencialmente conflictivas. Para añadir soporte a un backend de Terraform basado en bucket, crea un bucket en tu entorno cloud y luego busca y reemplaza todas las referencias del código a doit-matt-tf-state-test por el nombre de tu bucket.
Con el bucket de backend de Terraform listo, ya puedes levantar la infraestructura.
Si vas a correr tu workload en Google Cloud, ve a GCP/terraform. Dentro de esta carpeta hay subcarpetas etiquetadas standard y autopilot, cada una con código TF que lanzará un cluster de GKE en modo Standard o Autopilot. Autopilot suele ser la opción preferida porque no solo lleva los nodos (recursos de cómputo) hacia un modelo serverless, sino que además impone el uso de muchas mejores prácticas de seguridad.
Lamentablemente, Autopilot no funcionará en esta demo por una limitación: el espacio efímero en disco está limitado a 10 GiB, lo cual no alcanza para correr FastQC ni bwa-mem2 sobre datos reales. De todos modos, si esta limitación se elimina en el futuro, se recomienda usar Autopilot por sobre Standard. Dejé código TF para Autopilot por si esta restricción se levanta tras la publicación. AWS ECS Fargate también tiene un límite de 200 GiB en disco, insuficiente para la mayoría de los usos reales de cómputo científico. El código TF de Fargate no se incluye en el repositorio.
Si vas a correr tu workload en AWS, ve a AWS/terraform.
Una vez dentro de la carpeta de terraform específica de tu nube, ejecuta lo siguiente:
terraform apply -auto-approve# Ejecuta primero 'terraform plan' para ver la lista de cambios planeados sin crear nada
Esto planificará y luego ejecutará de inmediato el conjunto de archivos ‘.tf’ presentes en la carpeta para levantar un cluster de K8s y sus recursos prerrequisito. Terraform pedirá un GCP Project ID o un AWS Account ID, y nada más.
Opcionalmente puedes editar el archivo variables.tf antes de correr apply si quieres cambiar la región donde se lanzará el cluster, su nombre, dejar fijo el ID del proyecto GCP / cuenta AWS, etc.
Te recomiendo encarecidamente revisar el código para aprender Terraform
Para acelerar tu comprensión, en GCP se crean los siguientes recursos:
- Habilitación de las APIs de Compute, Container e IAM
- Un cluster GKE Standard con autoescalado dentro de la VPC por defecto, usando la versión de Kubernetes por defecto y actualizada (v1.20 al momento de publicación), con algunas funciones opcionales de mejores prácticas de seguridad habilitadas, como Workload Identity, Shielded Nodes y Secure Boot. Es un cluster regional con nodos repartidos en múltiples zonas (centros de datos)
- Una Service Account (SA) de GCP y su Kubernetes Service Account (KSA) vinculada que permiten a Workload Identity acotar el alcance de los permisos GCP para todos los jobs ejecutados dentro de cierto namespace de Kubernetes. La SA/KSA podrá leer/escribir en buckets y enviar varios logs y métricas a Cloud Logging y Cloud Metrics
- El cluster usa instancias preemptibles / Spot. Pagarás bastante menos que el precio on-demand, con la posibilidad de que los nodos caigan inesperadamente y eventualmente sean reemplazados. K8s identificará automáticamente los jobs fallidos por la terminación repentina de un nodo y los volverá a programar
Para AWS, se crean los siguientes componentes de red y seguridad:
- Una nueva VPC dedicada a alojar el cluster de K8s
- Subredes públicas (sin uso) y privadas + tablas de rutas para los nodos del cluster
- Un NAT Gateway, endpoints VPC para S3 y ECR con sus security groups asociados con TLS habilitado, además del soporte de DNS hostname para habilitar un cluster de nodos privados
- Un Internet Gateway para habilitar el acceso público al control plane del cluster
Para AWS, también tendrás que ir a AWS/terraform/ecr y volver a ejecutar terraform apply, ya que los archivos .tf de esta carpeta crean:
- Los repositorios donde se almacenarán las imágenes de contenedor que crees
(En GCP no es necesario crear los repositorios)
Para AWS, también deberás ir a AWS/terraform/eks y volver a ejecutar terraform apply, ya que los archivos .tf de esta carpeta crean:
- Un rol IAM para el cluster que habilita acceso de lectura y escritura a buckets S3, además de SSH sin claves a los nodos vía navegador a través de SSM Manager
- Un cluster EKS con autoescalado usando la versión de Kubernetes por defecto y actualizada (v1.21 al momento de publicación). Workload Identity, Shielded Nodes y Secure Boot no están disponibles fácilmente en EKS. Es un cluster regional con nodos repartidos en múltiples zonas (centros de datos)
- El cluster usa instancias preemptibles / Spot. Pagarás bastante menos que el precio on-demand, con la posibilidad de que los nodos caigan inesperadamente y eventualmente sean reemplazados. K8s identificará automáticamente los jobs fallidos por la terminación repentina de un nodo y los volverá a programar
Una vez completadas las operaciones de terraform apply, tu cluster está operativo y listo para que le envíes comandos de la API de Kubernetes mediante kubectl. Solo autentica kubectl con un comando específico de la nube:
# Para GCP:
gcloud container clusters get-credentials --region=us-central1 bioinformatics-tasks
# Para AWS:
aws eks update-kubeconfig --name=bioinformatics-tasks
Ya podrás ejecutar varios comandos contra el cluster, como listar los pods desplegados en todos los namespaces con kubectl get pods -A:
Pods típicos en un cluster GKE Standard nuevo y totalmente gestionado
Con el cluster levantado, ahora hay que instalar argo, ya que nos permitirá enviar no solo tareas individuales, sino también un workflow / pipeline de tareas dirigido por DAG.
Para hacerlo en GCP, ve a GCP/terraform/standard/kubectl_commands
Para hacerlo en AWS, ve a AWS/terraform/kubectl_commands
Ejecutar terraform apply -auto-approve en estas carpetas correrá comandos kubectl gestionados por Terraform que:
- Crean un namespace ‘biojobs’ al que se enviarán los jobs y (en GCP) acotan el alcance de permisos solo a leer/escribir objetos desde un bucket.
- Instalan argo y, en GCP con su Workload Identity, le otorgan permisos correctamente acotados para monitorear el progreso de los pods en el namespace ‘biojobs’.
Puedes verificar que el namespace se creó y que argo se instaló listando todos los namespaces y luego consultando los pods que corren en el namespace argo:
kubectl get ns
Todos los namespaces del cluster. EKS también tendrá ‘amazon-cloudwatch.’
kubectl get pods -n argo
Pods del servidor de Argo y del workflow controller corriendo en el namespace ‘argo’
Por último, ejecuta este comando para asegurarte de que argo se lleve bien con el cambio introducido en Kubernetes ≥v1.19, que pasó de usar docker a containerd como runtime de contenedores. No te preocupes mucho por este detalle, ya que argo terminará resolviéndolo:
kubectl patch configmap workflow-controller-configmap -n argo --patch ‘{"data":{"containerRuntimeExecutor":"k8sapi"}}’
Con argo instalado y los nodos de cómputo listos, ¡manos a la obra!
Paso 3: Construye las imágenes de contenedor de FastQC y bwa-mem2
Para correr workloads de FastQC y bwa-mem2 en AWS o GCP, hay que construir imágenes de contenedor para estas herramientas y almacenarlas en el repositorio de imágenes de contenedor de tu nube respectiva (GCR para GCP, ECR para AWS).
Ve a images/fastqc/
Dentro de esta carpeta está el Dockerfile que define cómo empaquetar FastQC en un contenedor junto con s5cmd (una herramienta para copias rápidas AWS S3 ←→ local) y gsutil (para copias GCS ←→ local). Tómate un tiempo para revisar el contenido del archivo y leer cómo se construyen los Dockerfiles. Como tema relacionado, te puede interesar este artículo que escribí sobre cómo maximizar la velocidad de transferencia de datos hacia buckets, ya que el Dockerfile aplica principios tomados de él.
Ejecuta build_fastqc_image_gcp.sh o build_fastqc_image_aws.sh para crear una imagen de contenedor a partir del Dockerfile y luego subirla a GCR o ECR, respectivamente.
Para crear una imagen de contenedor para bwa-mem y almacenarla en GCR o ECR, ve a images/bwa-mem2/ y repite el mismo proceso de ejecución del script de build.
Con este paso completo, ya tienes FastQC y bwa-mem2 disponibles para ejecutarse en un cluster de Kubernetes mediante las imágenes de contenedor almacenadas en el repositorio de imágenes de tu entorno cloud.
Paso 4: Envía y monitorea jobs de Kubernetes
A continuación iremos a la carpeta específica de tu nube para los jobs de Kubernetes y lanzaremos algunos. Más adelante correremos FastQC y bwa-mem2 en un pipeline con argo, pero para empezar, ejecutaremos estas tareas individualmente.
Para GCP, ve a GCP/gke/FastQC/
Para AWS, ve a AWS/eks/FastQC/
Dentro de estas carpetas hay un Job de Kubernetes llamado fastqc.yaml.
Este YAML define un job que se puede correr con kubectl create -f fastqc.yaml. Sin embargo, el archivo contiene algunas variables que deberían ser definidas y reemplazadas por run_fastqc_job.sh. Tómate un tiempo para entender los objetos de Kubernetes en general y los jobs en particular para descifrar fastqc.yaml, además de revisar lo que ocurre en el shell script.
En esencia, Kubernetes se usa con más frecuencia para correr pods de larga duración llamados Deployments (por ejemplo, servidores web de larga vida), pero también puede correr Jobs, o tareas de ejecución única, como programas científicos del estilo de FastQC. A lo largo del código, estaremos enviando objetos Job de Kubernetes al cluster.
Pásale al shell script las variables INPUT_FILEPATH y OUTPUT_FILEPATH y este lanzará un job de FastQC usando uno de los archivos FASTQ que descargaste antes, guardando finalmente el reporte comprimido en un bucket. Como alternativa, podrías reemplazar las variables del archivo fastqc.yaml manualmente y correr el job con: kubectl create -f fastqc.yaml
Sigue el progreso de FastQC con: kubectl get pods -n biojobs
Verás que se lanza un único pod, que primero descarga la imagen de contenedor y poco después corre el job especificado:

Pod de FastQC — estado ContainerCreating

Pod de FastQC — estado Running
Revisa los logs de la aplicación mientras corre con kubectl logs -n biojobs <pod_name>:
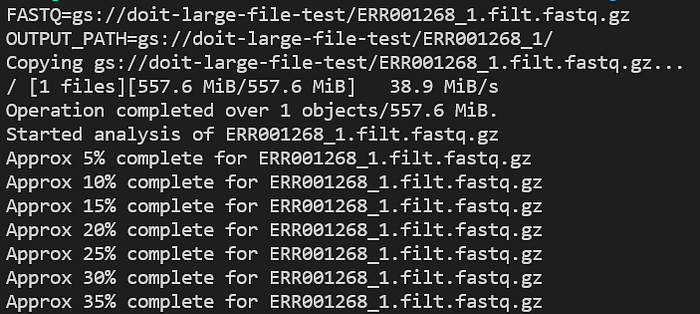
Progreso del pod de FastQC
Como los contenedores —y GKE/EKS— están construidos pensando en el monitoreo, puedes localizar fácilmente estas entradas de log en la solución de logging nativa de tu nube, lo que habilita retención a largo plazo y capacidades de investigación.
Usa la frase complete for en Cloud Logging de GCP o CloudWatch de AWS para localizar entradas de log de pods de FastQC:
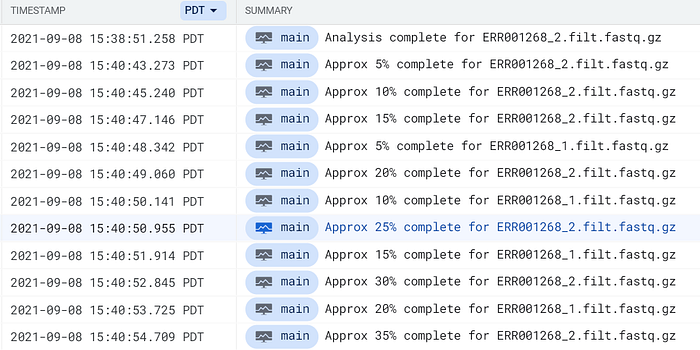
Logs de pods de FastQC llegando a Cloud Logging de GCP
Eventualmente verás aparecer un reporte de FastQC en la ruta de tu bucket de salida. Prueba correr pods de FastQC para ambos archivos FASTQ hasta ver sus reportes:
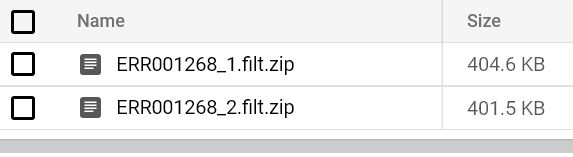
Reportes de FastQC almacenados en bucket
¡Eso es todo lo que hay que hacer para correr FastQC a escala en Kubernetes!
Podrías lanzar un job de FastQC o diez mil a la vez. El control plane de Kubernetes, junto con el servicio de autoescalado de nodos de cómputo asociado al entorno cloud, aumentará y disminuirá la capacidad según haga falta para cumplir con la demanda de los workloads, conforme a los requisitos de cpu/memoria/disco definidos en el archivo YAML del objeto Kubernetes de FastQC.
Solo asegúrate de que los valores máximos permitidos del autoscaler de tu cluster para cantidad de nodos / cpu / memoria sean lo suficientemente altos —o al menos donde los quieras para fines de control de costos— al momento de crear el cluster. (GCP: gke.tf AWS: cluster.tf)
¡Y a bwa-mem2 lo puedes correr a escala de manera costo-efectiva con un nivel de facilidad similar!
Para GCP, ve a GCP/gke/bwa-mem2/
Para AWS, ve a AWS/eks/bwa-mem2/
Provee rutas de entrada/salida para las variables al inicio de run_bwa-mem2_job.sh y estas reemplazarán los nombres de variable en bwa-mem2.yaml; luego envía el Job a tu cluster.
Sigue monitoreando el estado y los logs del pod hasta ver un archivo BAM en la ruta de salida de tu bucket:
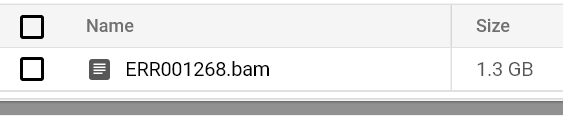
Archivo BAM almacenado en bucket
Si revisas las métricas de utilización de CPU de los nodos GCE / EC2 que dan vida a tu cluster, notarás que los jobs de FastQC, de baja intensidad y un solo core, se envían a máquinas con menos CPU, mientras que los jobs de BWA, que utilizan todos los cores, se envían a máquinas con alta CPU. Esto se corresponde con los grupos de nodos de Kubernetes de baja/alta CPU creados para asegurar que las máquinas adecuadamente dimensionadas, que dan vida a workloads tan distintos, escalen automáticamente hacia arriba y hacia abajo de manera costo-efectiva.
Por ejemplo, en AWS no tendría sentido aprovisionar una máquina m5.24xlarge de 96 vCPU al escalar solo para correr un nuevo job de FastQC de un solo core, así que en su lugar enviamos FastQC a un grupo de nodos con máquinas m5.xlarge de 4 vCPU. Por su parte, bwa requiere tantos cores como puedas darle, así que enviamos esos jobs al grupo de nodos de 96 vCPU, que escalará independientemente del grupo de 4 vCPU (FastQC).
Para quienes han construido workflows de análisis secundario que toman datos crudos de secuenciación y los procesan para descubrir variantes, probablemente ya estén empezando a ver el poder de Kubernetes. Atrás quedaron los días de:
- Schedulers de jobs on-prem con un rendimiento caprichoso a escala
- Preocuparte por si lanzar demasiados jobs a la vez tumbará al scheduler de jobs, o incluso al cluster que da vida a tus workloads (y los de los demás).
- Hacer enojar a tus colegas porque les quitas capacidad de cómputo on-prem que necesitan para correr sus workloads.
- Tener que monitorear y relanzar workloads que fallan por razones inesperadas pero recuperables.
- Quedarte sin pelo intentando rescatar archivos de log de jobs que crashearon — muchas veces de aplicaciones sin una funcionalidad de logging bien implementada.
Ahora, si tan solo no tuvieras que escribir código spaghetti para encadenar pasos de un pipeline…
Paso 5: Envía Workflows de Argo (Pipelines de Jobs de Kubernetes)
Argo te permite encadenar jobs en pipelines de manera que sigan operando a escala y soporten reintentos tanto de jobs como de pipelines ante fallos inesperados.
Veamos cómo usar argo para correr dos pods de FastQC simultáneamente. Después, solo si esos dos pods se completan con éxito, se lanza el alineamiento con bwa-mem2 sobre el mismo par FASTQ.
(Como ejercicio personal: podrías considerar añadir un paso de pass/no-pass a la imagen de FastQC, de modo que si el FASTQ tiene una calidad lo suficientemente baja como para no valer la pena alinearlo, se omita el paso de BWA en este workflow de argo.)
Para GCP, ve a GCP/gke/argo_workflow/
Para AWS, ve a AWS/eks/argo_workflow/
Provee rutas de entrada/salida para las variables al inicio de run_fastqc_to_bwa.sh y estas reemplazarán los nombres de variable en fastqc_to_bwa.yaml; luego envía el Job a tu cluster.
Ejecuta continuamente kubectl get pods -n biojobs y verás cómo corren los pods de FastQC, luego se completan, seguidos por el lanzamiento de un job de BWA:

Pods de FastQC corriendo sobre los archivos FASTQ R1 y R2 de NA12878
…
Pod de alineamiento bwa-mem2 levantado tras completarse los dos pods de FastQC
Echemos un vistazo al pod de bwa-mem2 con:
kubectl logs -n biojobs <pod_name> main
(Ten en cuenta que se agregó ‘main’ después del nombre del pod. Con argo, hay logs de pod ‘main’ y ‘wait’ para cada job dentro de un workflow.)
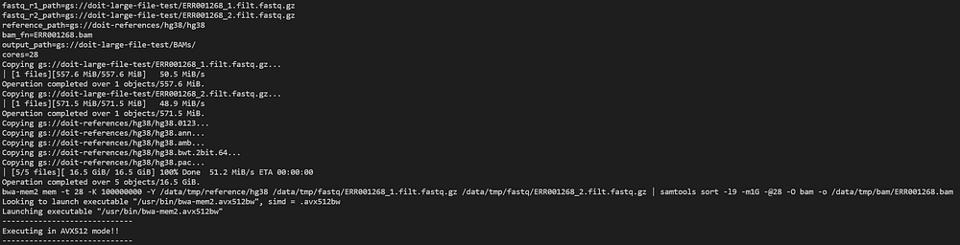
bwa-mem2 trabajando duro como parte de un workflow de argo
Puedes ejecutar kubectl get wf -n biojobs para monitorear el progreso del workflow de argo:

Eventualmente, cuando BWA termine, el workflow se habrá completado y encontrarás un par de reportes de FastQC y un BAM en tu bucket de salida.
Paso 6: Limpieza
Levantamos infraestructura de cómputo bastante cara para dar vida a BWA; no queremos este cluster GKE/EKS dando vueltas cuando no se está usando.
Simplemente recorre en orden inverso las carpetas de Terraform y ejecuta:
terraform destroy -auto-approve
Ten en cuenta que esto no eliminará los buckets que creaste manualmente para actuar como backend de Terraform y para almacenar los archivos FASTQ y de referencia. Terraform solo conoce y puede monitorear cambios en los recursos que él mismo levantó. En GCP, también tendrás que eliminar manualmente las imágenes de contenedor en GCR.
Para GCP, esto significa ejecutar el comando destroy en este orden:
GCP/terraform/standard/kubectl_commands/
GCP/terraform/standard/
Para AWS, esto significa ejecutar el comando destroy en este orden:
AWS/terraform/eks/kubectl_commands/
AWS/terraform/eks/
AWS/terraform/ecr/ (si quieres eliminar los repositorios de imágenes)
AWS/terraform/
Paso 7: Próximos pasos
Con las habilidades aprendidas hasta este punto, puedes construir pipelines de complejidad arbitraria que dependan de cualquier aplicación, ya que cualquier programa puede contenedorizarse.
Con código de imagen de contenedor versionado, contenedores con tags de versión, infraestructura versionada y pipelines de argo, puedes desplegar nuevas versiones de software y de pipelines, deprecar versiones antiguas y hacer rollback ante un despliegue defectuoso, con relativa facilidad — al menos comparado con las soluciones alternativas — si sigues por este camino de aprendizaje.
De ti depende decidir cuáles son los próximos pasos, pero espero haberte dejado con las habilidades centrales necesarias para la computación científica moderna y escalable.
Las mejores prácticas solo te llevan hasta cierto punto en la nube. Construir pensando en alta disponibilidad, resiliencia frente a fallos y escalabilidad optimizada en costos es hoy enormemente desafiante. Estas tareas adicionales de ingeniería se suman a una pila de workloads en constante crecimiento, algo que impacta directamente en la productividad.
Como científico en el siglo XXI, es esencial construir aplicaciones pensando en escalabilidad y facilidad de despliegue. No hacerlo solo trae dolores de cabeza para tu organización. A largo plazo, estos problemas se convierten en una bola de nieve de cuestiones serias e interminables en producción.
Si este proyecto te inspira, pero también te abruma un poco, ponte en contacto con DoiT International . También puedes escribirme por LinkedIn .
En DoiT nos enorgullece guiar a nuestros clientes hacia arquitecturas cloud optimizadas en costos, escalables, de alta disponibilidad, resilientes y preparadas para big data, un servicio que ofrecemos literalmente sin costo. Recibimos el premio Global Reseller Partner of the Year 2020 de Google Cloud con razón.
Conseguimos la distinción de certificación de la Partner Network (APN) de Amazon, manteniéndonos a la vez como AWS Advanced Consulting Partner, gracias a un talento de ingeniería de primer nivel y a un sólido soporte al cliente. Toda la información se te brinda on-demand para responder preguntas orientadas a la nube o resolver problemas en curso.
Muchas gracias por tu tiempo. Espero que estos dos artículos te hayan ayudado a mejorar tus habilidades. ¿Estás escalando? Contáctanos y déjanos ayudarte a alcanzar tus objetivos.
¡Gracias por leer! Para mantenerte conectado, síguenos en el DoiT Engineering Blog , el canal de LinkedIn de DoiT y el canal de Twitter de DoiT . Para explorar oportunidades de carrera, visita https://careers.doit-intl.com .