Internet adore les chats, donc je me dis que Cloud va m'attirer plus de vues que tous mes autres articles de blog réunis.
Quand ma femme m'a offert un kit Basepaws Whole Genome Sequencing pour notre cher chat Cloud à Noël dernier, j'étais aux anges. Pas seulement parce qu'il est franchement bluffant qu'une telle technologie existe et soit commercialement viable, mais aussi parce que, étant tellement habitué à analyser des données génomiques humaines, je peux apprécier — en tant que bio-informaticien — à quel point il est difficile de découvrir et de produire des résultats véritablement pertinents à partir d'un génome relativement peu étudié comme celui de Felis catus.
J'étais curieux de voir dans quelle mesure cette entreprise pouvait fournir des informations intéressantes ou exploitables sur mon animal, et dans quelle mesure je pourrais moi-même tirer quelque chose d'utile des données après avoir effectué mes propres analyses sur les données brutes de séquençage. La tâche n'a rien de simple : pour mener des analyses génomiques de manière rentable, il faut enchaîner en pipeline de nombreux outils complexes, et l'exécution de chaque étape doit être orchestrée sur des machines de tailles et de capacités variées. Qui suis-je pour refuser un tel défi pour les beaux yeux de ma petite boule de poils ?
Avant d'aborder la façon dont j'ai exploité la dernière version de Cloud Composer (le service Apache Airflow entièrement managé de GCP) pour simplifier l'exécution automatisée et la visualisation de plusieurs étapes complexes d'analyse génomique, ainsi que la façon dont j'ai utilisé Claude 3.5 Sonnet sur AWS Bedrock pour analyser les résultats, je tiens à souligner à quel point j'ai été impressionné par le rapport PDF généré par Basepaws.
Avec ses 68 pages, il contient bien plus que ce que je peux couvrir dans cet article. En résumé, le rapport présente clairement, pour votre animal :
- Les données de groupe racial (un échantillon est présenté ci-dessous)
- La présence ou l'absence de marqueurs génétiques de santé et de troubles génétiques
- Une multitude de marqueurs de traits intéressants
- Et, à ma grande surprise, un rapport de santé bucco-dentaire fondé sur le microbiome buccal
Le rapport sur la santé bucco-dentaire était particulièrement intéressant : il a confirmé certains problèmes déjà connus (par exemple, un risque moyen de maladie parodontale) et confirmé l'efficacité des mesures prises pour atténuer d'autres problèmes (par exemple, un risque faible de résorption dentaire, un trouble probablement d'origine partiellement génétique dont il souffre depuis des années et que mes vétérinaires ont su prendre en charge avec succès).
Analyser le génome de chat, déjà relativement peu annoté, est une chose ; séquencer en plus le microbiome buccal et en tirer des analyses pertinentes et exploitables en est une autre, et c'est un véritable accomplissement. Bravo à l'équipe scientifique qui a rendu cela possible.

J'avais envisagé de prendre un Maine Coon comme prochain chat — je n'aurais jamais deviné que j'en avais déjà (un peu) un !
Ceci dit, voyons ce que vous pouvez faire avec les données brutes du génome complet fournies par cette entreprise en complément de leur rapport.
Le dépôt lié ci-dessous, associé à cet article, exploite la version 3 récemment publiée de Cloud Composer et plusieurs autres services GCP de pointe — le tout déployé via le Terraform fourni — pour vous aider à automatiser, mettre à l'échelle et visualiser intégralement l'exécution de la trame complexe d'outils nécessaires à l'analyse secondaire et tertiaire des données du génome complet de chat.
https://github.com/doit-mattporter/genomics-workflow-orchestration
Pour aborder l'exécution d'analyses génomiques félines sur GCP, commençons par poser les bases de ce que ce dépôt va exécuter.
Pour ceux qui ne connaissent pas la bio-informatique : de manière générale, l'analyse de données génomiques se fait en trois grandes étapes de pipeline, quel que soit l'organisme étudié. L'analyse primaire est déjà réalisée au moment où vous recevez vos données de séquençage de Basepaws, tandis que les analyses secondaire et tertiaire sont exécutées à l'aide du code que je fournis. Voici ces trois étapes :
- Analyse primaire. Cette étape se déroule dans le centre de séquençage d'ADN et consiste à convertir les données de signal brutes (données lumineuses) issues des séquenceurs en séquences de paires de bases d'ADN (séquences A/C/G/T). Elle inclut également des contrôles QC, comme le retrait des appels de séquence à faible confiance.
En raison de limites techniques, il est impossible de séquencer un brin d'ADN d'un bout à l'autre en une seule lecture continue. Les brins d'ADN doivent au préalable être découpés en millions de petits fragments, qui sont ensuite seulement séquençables. La sortie de l'analyse primaire correspond donc à des millions — voire des milliards — de courts fragments d'ADN, longs en général de 100 à 300 paires de bases (pb). Ces données brutes et fragmentées vous sont fournies au format FASTQ, un format texte brut composé de chaînes de paires de bases ACGT pour chacune de ces millions de courtes séquences. C'est ce que BasePaws vous envoie, et c'est le point de départ de cet article et du dépôt associé. 2. Analyse secondaire. Cette étape revient à reconstituer un puzzle géant de votre ADN, dont la solution est toujours un peu différente de celle illustrée sur la boîte. Elle comporte deux composantes majeures :
a) " Alignement de séquences ", ou assemblage du puzzle ADN : les millions de courts fragments d'ADN issus de l'analyse primaire sont reconstitués — au prix d'une grande puissance de calcul — pour reformer le génome de chat original, intégralement reconstruit, dont ces fragments sont issus.
b) " Variant Calling ", ou jeu des sept différences : une fois le génome de votre chat réassemblé, il faut le comparer au génome de " référence " que la communauté scientifique considère comme le standard d'un génome de chat typique. Votre chat s'écartera de ce standard de " référence " du fait de son unicité génétique ; cette étape consiste à repérer ces différences génétiques, là où l'ADN de votre chat diverge de l'ADN de chat " typique ". Ces différences sont appelées " variants " ou " mutations ". Toutes les mutations détectées lors du variant calling ne sont pas réelles — beaucoup sont des erreurs, pour des raisons techniques complexes. Cette étape inclut donc également un QC, afin de s'assurer que les variants identifiés sont bien réels. 3. Analyse tertiaire. Souvent la partie la plus délicate et la plus cruciale, elle consiste à découvrir ce que la liste de différences ADN issue de l'analyse secondaire signifie réellement. L'analyse tertiaire correspond à la présentation de résultats tangibles et porteurs de sens. Certains de ces variants sont-ils associés à des problèmes de santé connus ? Comment mon chat va-t-il réagir à différents médicaments selon sa liste de mutations ? Telle mutation est-elle responsable de la couleur de son pelage ? Cet ensemble de mutations indique-t-il que mon chat est probablement d'ascendance Maine Coon ? L'analyse tertiaire permet de répondre à des questions de cette nature.
Maintenant que vous avez une idée assez claire du travail à accomplir, passons à la pratique ! Une fois que vous avez reçu les données du génome complet de votre chat de la part de BasePaws — ou si vous voulez simplement suivre le fil et voir ce qu'il est possible de faire avec Cloud Composer et Claude à partir de ces données —, vous pouvez procéder comme suit.
Le README du dépôt explique comment commencer par provisionner l'infrastructure cloud requise via une opération terraform apply. Notez que certaines ressources, notamment Cloud Composer, fonctionneront 24h/24 et 7j/7 jusqu'à leur arrêt : surveillez donc votre dépense cloud et veillez à arrêter les ressources inutilisées. Les ressources clés suivantes seront déployées :
Environnement Google Composer
- Environnement Composer 3 avec Apache Airflow 2
- Configure un DAG Airflow pour le traitement des données génomiques félines. Notez que ce DAG provisionnera temporairement des instances Compute Engine c4-standard-96 et c4-standard-8 selon les besoins des différentes étapes du pipeline.
Buckets Google Cloud Storage
genomic_inputs,genomic_reference,genomic_outputsetcloud_functionpour stocker respectivement les entrées (fichiers FASTQ), les génomes de référence, les sorties (VCF et fichiers d'annotation) et le code Cloud Function
Google Cloud Function v2
- Fonction
genomic-dag-kickoffv2 pour déclencher le DAG d'analyse génomique - Pilotée par événement à la finalisation d'un objet GCS. Cette fonction recherche un fichier
ready.txt, puis utilise la liste des emplacements de buckets FASTQ qu'il contient pour lancer le DAG sur ces FASTQ.
Instance Google Compute Engine
grabbing-reference-genomeest une instance c4-standard-96 qui télécharge deux génomes de référence félins depuis NCBI, indexe ces références avec GATK, puis téléverse les génomes de référence ainsi traités dans le bucketgenomic_referencepour une utilisation ultérieure par le DAG.- Notez que cette ressource n'a besoin d'être exécutée qu'une seule fois et s'auto-termine une fois son travail terminé.
Une fois ces ressources opérationnelles via terraform apply, il vous suffit de procéder comme suit pour lancer les analyses génomiques félines :
- Téléversez les FASTQ BasePaws dans le bucket
genomic_inputs_<random_id> - Créez un fichier
ready.txtcontenant les URI GCS de ces FASTQ et téléversez-le dans le même bucket.
Le téléversement du fichier ready.txt déclenchera la Cloud Function genomic-dag-kickoff. Celle-ci lancera deux instances de DAG sur Cloud Composer, chacune orchestrant l'analyse secondaire et tertiaire sur l'une des deux dernières versions du génome félin : Felis_catus_9.0 et Felis_catus_Fca126_mat1.0.
Le DAG exécute une orchestration complexe d'outils bio-informatiques qui produit au final :
- Des fichiers VCF, ou Variant Call Files. Il s'agit de la liste de toutes les mutations identifiées.
- Des fichiers d'annotation produits par SnpEff. SnpEff fournit des annotations in silico pour chaque mutation située dans un gène codant pour une protéine, en estimant la probabilité que la mutation entraîne un impact fonctionnel faible, modéré ou élevé sur la protéine codée par ce gène.
À haut niveau, le DAG est composé des Task Groups suivants : les Task Groups " alignment " et " variant calling " correspondent à l'analyse secondaire, et le Task Group " annotation " à l'analyse tertiaire :
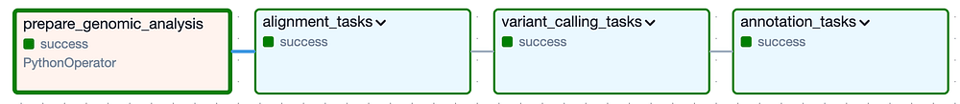
DAG de Cloud Composer pour l'exécution de l'analyse secondaire et tertiaire sur une version donnée du génome félin
En dépliant chacun des Task Groups (illustrés ci-dessous), on découvre la multitude de tâches individuelles nécessaires pour exécuter ce pipeline d'outils, puis pour vérifier que les fichiers de sortie attendus ont bien été créés. Notez que j'ai prévu une logique permettant d'ignorer entièrement chaque Task Group si ses fichiers de sortie sont déjà présents dans le bucket de sortie suite à une exécution précédente réussie.
Sans Apache Airflow et la version entièrement managée par GCP de cet outil open-source, enchaîner ces outils en pipeline et vérifier leur bonne exécution (ou leur exécution réussie antérieure) à chaque étape serait nettement plus complexe et chronophage à mettre en œuvre. Au-delà de l'orchestration de ces étapes, il faudrait aussi récupérer et stocker leurs logs, visualiser la progression du pipeline en temps réel et remonter le moindre incident.
Composer facilite la visualisation de la progression, la création de pipelines avec des branchements conditionnels, et garantit que les erreurs sont capturées et leurs logs présentés dans une UI facile à parcourir. Pour ces raisons et bien d'autres — comme la capacité de Composer à mettre à l'échelle les ressources de calcul de manière rentable dans des opérations à l'échelle de l'entreprise —, je considère les outils d'orchestration de workflow comme Composer essentiels à la construction de pipelines de données scalables.
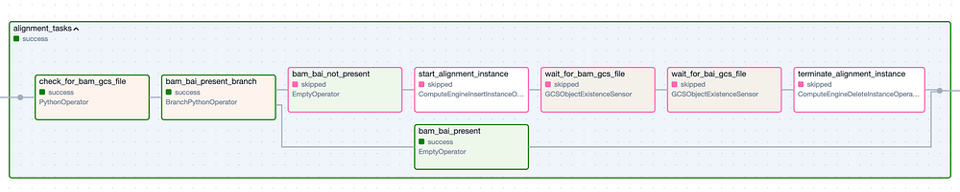
Le Task Group " Alignment ". Les étapes à bordure verte correspondent aux exécutions réussies, tandis que les étapes à bordure rose ont été ignorées.
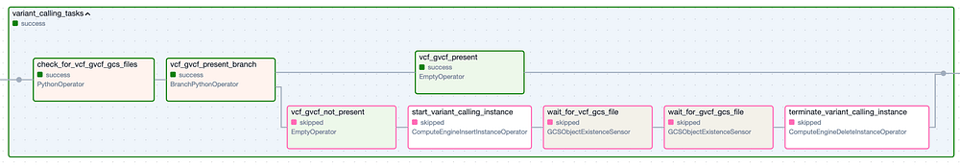
Le Task Group " Variant Calling ". Les étapes à bordure verte correspondent aux exécutions réussies, tandis que les étapes à bordure rose ont été ignorées.
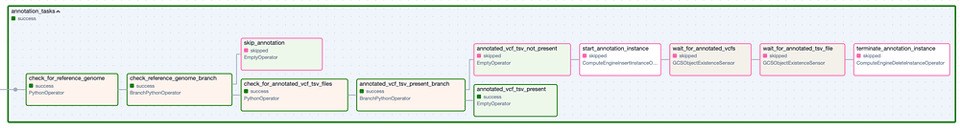
Le Task Group " Annotation ". Les étapes à bordure verte correspondent aux exécutions réussies, tandis que les étapes à bordure rose ont été ignorées.
Une fois toutes ces étapes terminées, vous verrez les VCF et les fichiers d'annotation déposés dans le bucket GCS genomic-outputs-<random_id>. Les VCF contiennent la liste de toutes les mutations identifiées, tandis que les fichiers d'annotation détaillent les effets prédits in silico par SnpEff de ces mutations sur les protéines codées par les gènes.
Ce qui nous intéresse ici, c'est précisément la façon dont SnpEff estime l'impact probable sur les gènes codant pour des protéines.
C'est à ce moment-là que j'ai vraiment commencé à mesurer la valeur du service BasePaws. Pour la génomique humaine, il existe une richesse étonnante de sources de données d'annotation de variants en open-source dans lesquelles puiser pour acquérir une compréhension clinique approfondie de pratiquement n'importe quelle mutation survenant dans un gène codant pour une protéine. À l'inverse, dans le domaine de la génomique féline, je n'ai pas réussi à trouver la moindre base d'annotation open-source, pas même pour les mutations aux conséquences cliniques claires et documentées. N'ayant guère envie de scraper des publications académiques pour me constituer ma propre base, il ne me restait plus qu'à intégrer SnpEff dans l'étape d'annotation de mon DAG Cloud Composer, ce qui nous donnera au moins des prédictions d'effet fonctionnel in silico sur lesquelles nous appuyer. BasePaws a sans doute consenti des efforts considérables pour bâtir des bases d'annotation en interne, afin d'offrir le type d'analyses qu'on retrouve dans le rapport qu'ils m'ont fourni.
Bref, déterminé à tirer parti des prédictions d'effet de SnpEff, je me suis tourné vers les LLM, et plus précisément Claude 3.5 Sonnet sur AWS Bedrock.
J'ai commencé par exécuter les commandes bash suivantes sur le fichier d'annotation produit par SnpEff. Ces commandes simples filtrent les 13,5 millions de mutations annotées pour n'en retenir que 366, plus susceptibles d'être " intéressantes " — c'est-à-dire celles qui :
- Sont situées dans une région codante pour une protéine
- Ont plus de chances d'être de véritables variants plutôt qu'un appel de variant erroné
- Possèdent un homologue humain identifié (un LLM aura plus de chances d'en saisir l'impact potentiel s'il connaît son équivalent protéique humain), et
- Présentent un impact prédit " ÉLEVÉ " sur l'effet fonctionnel d'une protéine
head -n1 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff.ann.tsv > 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact.ann.tsv
grep "\tHIGH\t.*protein_coding" 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff.ann.tsv >> 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact.ann.tsv
grep -v "frameshift" 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact.ann.tsv |
grep -v "intron" |
awk '$11 !~ /^ENSFCAG000/' > 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact_filtered.ann.tsv
cut -f 1,2,4,5,8-11,17,18,19,21 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact_filtered.ann.tsv
J'ai collé le contenu de ce fichier filtré, contenant 366 variants annotés à impact " ÉLEVÉ ", dans Claude, accompagné du prompt suivant :
Analysez les annotations génétiques SnpEff suivantes pour Felis catus 9.0. Lorsque vous faites référence à un gène, veillez à mentionner combien de mutations ont été identifiées dans ce gène et n'inventez pas de mutations pour des gènes qui ne figurent pas dans le fichier d'annotation collé ci-dessous. Veuillez fournir :
1. Un résumé des résultats les plus significatifs
2. Mettez en évidence toutes les mutations dans des gènes associés à des conditions de santé connues, des traits phénotypiques ou des processus biologiques importants.
3. Expliquez les implications fonctionnelles potentielles des 5 à 10 mutations les plus intéressantes, en tenant compte de la fonction du gène et du type de mutation.
4. Identifiez tout schéma, regroupement de mutations ou voies biologiques/fonctionnelles impactées qui mériteraient d'être notés.
5. Suggérez tout problème de santé potentiel ou trait susceptible d'être affecté en fonction de ces mutations, mais précisez qu'il s'agit de spéculations sans corrélation clinique.
Veuillez utiliser une terminologie scientifique lorsque c'est pertinent, mais fournissez également des explications accessibles aux profanes pour assurer la compréhension. Si vous avez besoin de précisions ou d'informations supplémentaires sur le phénotype ou les antécédents de santé du sujet pour affiner votre analyse, n'hésitez pas à demander.
Le chat a un pelage entièrement blanc, a 11 ans et n'a aucun problème de santé connu en dehors de la résorption dentaire féline. Le chat n'a aucun trouble neurologique ou immunitaire connu (hormis le lien possible entre la fonction du système immunitaire et la résorption dentaire féline).
Cela a donné les résultats suivants, que j'ai trouvés assez intéressants : Claude évoque effectivement une possible prédisposition génétique à la résorption dentaire féline en s'appuyant sur certains des gènes présentant des mutations à fort impact. Avec un pelage entièrement blanc, il est utile de savoir qu'il pourrait également être plus exposé au cancer en raison de la fonctionnalité potentiellement altérée d'une protéine de réparation de l'ADN, car cela signifie que je devrais limiter son temps d'exposition au soleil direct.
Les autres conclusions de Claude sont en revanche assez spéculatives, car je n'ai constaté aucun des problèmes graves rapportés concernant la fonction musculaire ou le métabolisme. Comme il n'existe pas de jeux de données d'annotation publiquement disponibles et bien organisés, la fonction des gènes félins est largement déduite par homologie avec leurs équivalents humains. Je m'attends donc à ce que les LLM fournissent des inférences fonctionnelles larges et centrées sur l'humain, d'où le caractère excessivement spéculatif des résultats produits par Claude.
Merci pour ces informations génétiques détaillées concernant votre chat blanc de 11 ans atteint de résorption dentaire féline. Je vais analyser les mutations à fort impact à partir des annotations SnpEff que vous avez partagées. Reprenons cela section par section :
1. Résumé des résultats les plus significatifs :
Les données génétiques révèlent un nombre substantiel de mutations à fort impact dans divers gènes. Ces mutations comprennent des variants stop-gained, start-lost et stop-lost, susceptibles d'altérer significativement la fonction des protéines. Au total, plus de 200 mutations à fort impact ont été identifiées dans de nombreux gènes.
2. Mutations dans des gènes associés à des conditions ou traits connus :
a) LRRC45 (1 mutation) : associé à la fonction ciliaire, qui peut affecter divers processus physiologiques.
b) PNKP (1 mutation) : impliqué dans la réparation de l'ADN, avec un impact potentiel sur la sensibilité au cancer et la fonction neurologique.
c) ATP2A1 (1 mutation) : crucial pour la fonction musculaire, en particulier dans les fibres musculaires à contraction rapide.
d) COQ7 (1 mutation) : important pour la biosynthèse de la coenzyme Q10, avec un impact sur la production d'énergie cellulaire.
e) ABCC6 (1 mutation) : associé à des troubles du tissu conjonctif chez l'humain.
f) CRP (1 mutation) : impliqué dans la réponse immunitaire et l'inflammation.
3. Implications fonctionnelles potentielles des principales mutations :
a) PNKP (c.1150C>T, p.Gln384*) : cette mutation stop-gained pourrait conduire à une protéine tronquée, ce qui altérerait potentiellement les mécanismes de réparation de l'ADN. Cela pourrait accroître la sensibilité aux dommages de l'ADN et, en théorie, affecter la fonction neurologique ou le risque de cancer.
b) ATP2A1 (c.3149A>G, p.Ter1050Trpext*?) : cette mutation stop-lost pourrait entraîner une protéine étendue. ATP2A1 est crucial pour la contraction musculaire, et de telles altérations pourraient potentiellement affecter la fonction musculaire.
c) COQ7 (c.817T>A, p.Ter273Lysext*?) : autre mutation stop-lost, elle pourrait également conduire à une protéine étendue. COQ7 est vital pour la production de coenzyme Q10, essentielle à la production d'énergie cellulaire. Des altérations pourraient affecter le métabolisme énergétique global.
d) ABCC6 (c.4246C>T, p.Arg1416*) : cette mutation stop-gained pourrait entraîner une protéine tronquée. Chez l'humain, les mutations d'ABCC6 sont associées au pseudoxanthome élastique, une affection du tissu conjonctif.
e) CRP (c.592T>C, p.Ter198Glnext*?) : cette mutation stop-lost dans le gène de la protéine C-réactive pourrait affecter la réponse inflammatoire et la fonction immunitaire.
4. Schémas et observations notables :
On observe un regroupement de mutations affectant des gènes impliqués dans la fonction musculaire, le métabolisme énergétique et la réponse immunitaire. Plusieurs mutations se trouvent dans des gènes liés à la réparation de l'ADN et aux processus de maintenance cellulaire.
5. Problèmes de santé ou traits potentiellement affectés (spéculatif) :
Sur la base de ces mutations, des préoccupations potentielles pourraient porter sur :
\* La fonction musculaire et le métabolisme (ATP2A1, COQ7)
\* La régulation du système immunitaire (CRP)
\* La réparation de l'ADN et la sensibilité potentielle au cancer (PNKP)
\* La santé du tissu conjonctif (ABCC6)
Cependant, il est essentiel de souligner que la présence de ces mutations ne signifie pas nécessairement que le chat développera les pathologies associées. De nombreux facteurs, dont les influences environnementales et d'autres modificateurs génétiques, jouent un rôle dans l'expression des gènes et le développement des maladies.
Il est intéressant de noter que, malgré ces mutations, votre chat a vécu jusqu'à 11 ans sans problème de santé majeur signalé en dehors de la résorption dentaire féline. Cela illustre la relation complexe entre génotype et phénotype.
Compte tenu des antécédents de résorption dentaire féline du chat, les mutations dans des gènes liés à la fonction immunitaire (comme CRP) et au tissu conjonctif (comme ABCC6) pourraient présenter un intérêt particulier, ces systèmes étant supposés jouer un rôle dans cette pathologie. Cependant, aucun lien direct ne peut être établi sans recherches supplémentaires.
Ensuite, j'ai voulu identifier les gènes connus pour être associés à un pelage blanc chez le chat, puis filtrer toutes les mutations annotées par SnpEff dans ces gènes, indépendamment de la prédiction d'impact (faible/modéré/élevé) de SnpEff. Cela a été obtenu en filtrant le fichier d'annotation pour les mutations dans les gènes KIT et KITLG :
head -n1 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff.ann.tsv > 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_LOW_MODERATE_impact_white_coat.ann.tsv
grep -E "\t(LOW|MODERATE)\t.*protein_coding" 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff.ann.tsv | grep -E '\tKIT\t|\tKITLG\t' >> 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_LOW_MODERATE_impact_white_coat.ann.tsv
cut -f 1,2,4,5,8-11,17,18,19,21 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_LOW_MODERATE_impact_white_coat.ann.tsv
En réutilisant le même prompt, Claude conclut que les mutations missense et de site d'épissage d'intron sont probablement à l'origine du pelage blanc de Cloud :
Merci d'avoir fourni les annotations génétiques pour Felis catus 9.0. Je vais analyser ces données et fournir les informations demandées. Reprenons les résultats :
1. Résumé des résultats les plus significatifs :
Les annotations révèlent plusieurs mutations dans deux gènes importants : KIT et KITLG. Ces gènes font partie de la voie de signalisation KIT/KITLG, qui joue un rôle crucial dans divers processus biologiques, notamment le développement de la couleur du pelage, l'hématopoïèse et la gamétogenèse.
Gène KIT : 9 mutations (7 variants synonymes, 2 variants de région d'épissage)
Gène KITLG : 2 mutations (1 variant missense, 1 variant synonyme)
Le résultat le plus significatif est le variant missense dans le gène KITLG, qui pourrait potentiellement affecter la fonction de la protéine.
2. Mutations associées à des conditions de santé connues, des traits phénotypiques ou des processus biologiques importants :
Les gènes KIT et KITLG sont associés à la couleur du pelage chez le chat, en particulier aux motifs de tachetage blanc. Le pelage entièrement blanc de ce chat pourrait être lié à ces mutations, surtout au vu du nombre de variants présents dans le gène KIT.
3. Implications fonctionnelles potentielles des 5 à 10 mutations les plus intéressantes :
a) Variant missense KITLG (c.731G>A, p.Ser244Asn) : cette mutation transforme une sérine en asparagine en position 244 de la protéine KITLG. Elle pourrait potentiellement affecter la fonction de la protéine, puisqu'elle modifie la séquence d'acides aminés. KITLG est crucial pour la survie et la prolifération des mélanocytes, qui produisent le pigment. Cette mutation pourrait contribuer au pelage entièrement blanc du chat.
b) Variants de région d'épissage KIT (c.2491–3C>T, c.1353–5T>C, c.1122–4_1122–3delGT) : ces mutations se produisent dans des régions d'épissage, importantes pour le traitement correct de l'ARNm. Bien que classées à faible impact, elles pourraient potentiellement affecter l'épissage du gène KIT, conduisant à une production ou une fonction protéique altérée. La protéine KIT est un récepteur tyrosine kinase impliqué dans divers processus de développement, dont celui des mélanocytes.
c) Variants synonymes KIT : bien que les variants synonymes ne modifient pas la séquence d'acides aminés, ils peuvent néanmoins influencer l'expression du gène par divers mécanismes, comme l'altération de la stabilité de l'ARNm ou de l'efficacité de la traduction. Le nombre élevé de variants synonymes dans le gène KIT (7 au total) pourrait collectivement influencer son expression ou sa fonction.
4. Schémas, regroupements de mutations ou voies biologiques impactées :
Les mutations sont regroupées dans la voie de signalisation KIT/KITLG, cruciale pour le développement et la fonction des mélanocytes. Cette voie est également impliquée dans l'hématopoïèse (formation des cellules sanguines) et la gamétogenèse (production des cellules reproductrices). La concentration de mutations dans ces gènes suggère un impact potentiel sur la pigmentation et, possiblement, sur d'autres processus apparentés.
5. Problèmes de santé ou traits potentiellement affectés (spéculatif) :
a) Couleur du pelage : le pelage entièrement blanc du chat est probablement lié à ces mutations, en particulier dans les gènes KIT et KITLG.
b) Audition : certains chats entièrement blancs porteurs de mutations KIT sont sujets à la surdité congénitale. Bien qu'aucune mutation spécifiquement associée à la surdité ne soit présente ici, il pourrait être utile de surveiller l'audition du chat.
c) Fonction immunitaire : la voie KIT/KITLG joue un rôle dans l'hématopoïèse, y compris dans le développement de certaines cellules immunitaires. Bien que le chat ne présente aucun trouble connu du système immunitaire, ces mutations pourraient subtilement influencer la fonction immunitaire.
d) Résorption dentaire : bien qu'elle ne soit pas directement liée à KIT/KITLG dans la littérature actuelle, l'implication de cette voie dans divers processus de développement pourrait indirectement contribuer à des problèmes dentaires. Ceci est hautement spéculatif et nécessiterait des recherches supplémentaires pour établir un lien éventuel.
e) Fertilité : compte tenu du rôle de KIT/KITLG dans la gamétogenèse, il pourrait y avoir des effets potentiels sur la fertilité. Cela reste cependant spéculatif et ne serait pertinent que si le chat devait être reproduit.
Il est important de noter que, si ces résultats génétiques sont intéressants, leur signification clinique ne peut être établie qu'en les corrélant à l'état de santé réel du chat et par des études complémentaires. Le bon état de santé actuel du chat à 11 ans suggère que beaucoup de ces mutations n'ont peut-être pas d'impact sévère.
Ce que je retiens de tout cela, c'est qu'il est possible de tirer des constats vraiment intéressants des données génomiques de votre chat, et que l'obtention de ces données est rendue nettement plus accessible, fiable et scalable grâce aux services cloud d'orchestration de pipelines de données comme Cloud Composer.
Cependant, beaucoup reste à faire du côté de la recherche financée sur fonds publics. Inférer la fonctionnalité des gènes félins à partir de leur homologie avec les gènes humains ne permet d'aller que jusqu'à un certain point en matière d'analyses pertinentes. Lorsque les institutions de recherche vétérinaire commenceront à publier des bases d'annotation génomique pour les chats et les chiens, je suis convaincu que les analyses génomiques alimentées par les LLM révéleront tout leur potentiel. En attendant, je reste impressionné par le travail scientifique mené par l'équipe Basepaws.
Et voilà ! Nous avons exploré la fascinante frontière de la génomique féline, du rapport impressionnant de BasePaws à notre propre analyse DIY portée par des services de cloud computing de pointe et l'IA générative. Orchestrer une symphonie d'outils génomiques peut être aussi délicat que de rassembler une portée de chatons, mais des services comme Cloud Composer rendent l'exercice nettement plus simple.
PerfectScale™ for Kubernetes
Ready to optimize?
Get your free Kubernetes savings analysis
Your cloud bill shouldn't be a mystery
Optimization, automation, expertise. In one platform.