Internet adora i gatti, quindi scommetto che Cloud mi farà guadagnare più visualizzazioni di tutti gli altri post del blog messi insieme.
Quando lo scorso Natale mia moglie mi ha regalato un kit Basepaws Whole Genome Sequencing per il nostro amato gatto Cloud, ero entusiasta. Non solo perché trovo straordinario che questa tecnologia esista e sia ormai accessibile sul mercato, ma anche perché sono talmente abituato ad analizzare dati genomici umani che, da bioinformatico, so bene quanto sia difficile individuare e descrivere risultati davvero significativi a partire da un genoma relativamente meno studiato come quello di Felis catus.
Ero curioso di scoprire fino a che punto questa azienda fosse in grado di fornire informazioni interessanti o utili sul mio animale, e fino a che punto io stesso potessi ricavare qualcosa di utile dai dati grezzi di sequenziamento eseguendo le mie analisi. Non è un compito banale: per portare a termine analisi genomiche in modo economicamente sostenibile occorre concatenare in pipeline molti strumenti complessi, e l'esecuzione di ogni passaggio va orchestrata su macchine di dimensioni e capacità diverse. E chi sono io per tirarmi indietro davanti a una sfida del genere, soprattutto per il mio adorabile micione?
Prima di raccontare come ho sfruttato l'ultima versione di Cloud Composer (il servizio Apache Airflow completamente gestito di GCP) per semplificare l'esecuzione automatica e la visualizzazione di vari passaggi complessi di analisi genomica, e come ho usato Claude 3.5 Sonnet su AWS Bedrock per analizzare i risultati, voglio partire sottolineando quanto mi abbia colpito il report PDF prodotto da Basepaws.
Con le sue 68 pagine, contiene molto più di quanto io possa trattare in questo articolo. In sintesi, però, il report comunica chiaramente, per il vostro animale:
- i dati relativi al gruppo di razza (un esempio è mostrato qui sotto);
- la presenza/assenza di marcatori genetici di salute e di disturbi genetici;
- una moltitudine di marcatori di tratti molto interessanti;
- e, con mia sorpresa, un report sulla salute orale basato sul microbioma del cavo orale.
Il report sulla salute orale è stato particolarmente interessante: ha confermato alcuni problemi già noti (ad esempio un rischio 'Medio' di malattia parodontale) e ha confermato l'efficacia delle azioni intraprese per mitigarne altri (ad esempio un rischio 'Basso' di riassorbimento dentale, un problema probabilmente in parte di origine genetica che il mio gatto ha da anni e che i veterinari hanno saputo gestire con successo).
Già di per sé è complicato analizzare il genoma felino, relativamente poco annotato, ma sequenziare anche il microbioma orale e ricavarne analisi significative e concretamente utilizzabili è un risultato notevole. Complimenti al team di scienziati che lo ha reso possibile.

Avevo pensato di prendere un Maine Coon come prossimo gatto: non avrei mai immaginato di averne già (più o meno) uno!
Detto questo, vediamo cosa si può fare con i dati grezzi dell'intero genoma che l'azienda fornisce insieme al report.
Qui sotto trovate il link al repo associato a questo blog, che sfrutta la versione v3 di Cloud Composer rilasciata di recente e altri servizi GCP all'avanguardia — tutti avviati con il Terraform fornito — per automatizzare, scalare e visualizzare completamente l'esecuzione della complessa rete di strumenti necessari per le analisi secondaria e terziaria sui dati dell'intero genoma felino.
https://github.com/doit-mattporter/genomics-workflow-orchestration
Cominciamo dunque a parlare di come eseguire analisi genomiche feline su GCP, partendo da una panoramica di base di ciò che il repo eseguirà.
Per chi non ha familiarità con la bioinformatica: in linea generale, esistono tre grandi fasi di pipeline che descrivono come si analizzano i dati genomici, indipendentemente dall'organismo studiato. L'analisi primaria sarà già stata effettuata quando riceverete i dati di sequenziamento da Basepaws, mentre le analisi secondaria e terziaria si eseguono con l'aiuto del codice che ho condiviso. Ecco le tre fasi:
- Analisi primaria. Questo passaggio avviene presso la struttura di sequenziamento del DNA e consiste nel convertire i dati grezzi del segnale (dati luminosi) prodotti dai sequenziatori in sequenze di coppie di basi del DNA (sequenze di A/C/G/T). Comprende inoltre controlli di QC, come la rimozione delle chiamate di sequenza a bassa affidabilità.
Per limiti tecnici, è impossibile sequenziare un filamento di DNA dall'inizio alla fine in un'unica lettura continua. I filamenti devono invece essere frammentati in milioni di pezzi corti, che solo a quel punto possono essere sequenziati. L'output dell'analisi primaria è quindi composto da milioni — se non miliardi — di brevi frammenti di DNA, ciascuno tipicamente lungo da 100 a 300 coppie di basi (bps). Questi dati di sequenziamento grezzi e frammentati vi vengono forniti in formato FASTQ, in pratica un formato di testo semplice composto da stringhe di valori ACGT delle coppie di basi per questi milioni di brevi sequenze. È ciò che BasePaws vi invia ed è il punto di partenza di questo blog e del repo associato. 2. Analisi secondaria. Questa fase è come comporre un puzzle gigantesco del vostro DNA, dove la soluzione è sempre un po' diversa dall'immagine sulla scatola. Si articola in 2 componenti principali:
a) 'Sequence Alignment', ovvero l'assemblaggio del puzzle del DNA: i milioni di brevi frammenti di DNA prodotti dall'analisi primaria vengono ricomposti — con un'enorme quantità di potenza di calcolo — per ricostruire il genoma felino originale e integro da cui provenivano.
b) 'Variant Calling', ovvero il gioco delle differenze: una volta riassemblato il genoma del vostro gatto, lo si confronta con il genoma di 'riferimento' che la comunità scientifica ha stabilito come gold standard per il genoma felino tipico. Il vostro gatto si discosterà da questo standard di 'riferimento' a causa della propria unicità genetica; questo passaggio consiste nell'individuare quelle differenze, i punti in cui il DNA del vostro gatto si scosta da quello felino 'tipico'. Tali differenze si chiamano 'varianti' o 'mutazioni'. Non tutte le mutazioni rilevate durante il variant calling sono reali: molte sono errori, per ragioni complesse. Anche questa fase prevede QC, per assicurarsi che le varianti siano effettivamente reali. 3. Analisi terziaria. Spesso la parte più delicata e cruciale, consiste nello scoprire cosa significhi davvero l'elenco di differenze del DNA emerso dall'analisi secondaria. L'analisi terziaria è la presentazione di risultati tangibili e significativi. Alcune di queste varianti sono note per essere associate a condizioni di salute? Come risponderà il mio gatto alle diverse opzioni farmacologiche in base al suo elenco di mutazioni? Questa specifica mutazione è responsabile del colore del suo mantello? Questo insieme di mutazioni indica che il mio gatto ha probabilmente ascendenza Maine Coon? L'analisi terziaria comprende la capacità di rispondere a domande di questo tipo.
Ora che avete un'idea sufficientemente chiara del lavoro da fare, mettiamoci all'opera! Una volta ricevuti da BasePaws i dati dell'intero genoma del vostro gatto — o se volete semplicemente seguire l'articolo per vedere cosa si può fare con Cloud Composer e Claude usando questi dati — potete procedere come segue.
Il README del repo descrive come iniziare effettuando il provisioning dell'infrastruttura cloud necessaria con un'operazione terraform apply. Attenzione: alcune risorse, in particolare Cloud Composer, restano attive 24/7 finché non le terminate, quindi tenete d'occhio la spesa cloud e ricordatevi di chiudere le risorse inutilizzate. Verranno avviate queste risorse principali:
Google Composer Environment
- Ambiente Composer 3 con Apache Airflow 2.
- Configura un DAG di Airflow per l'elaborazione dei dati genomici felini. Questo DAG effettua temporaneamente il provisioning di istanze Compute Engine c4-standard-96 e c4-standard-8, in base ai requisiti dei vari step della pipeline.
Google Cloud Storage Buckets
genomic_inputs,genomic_reference,genomic_outputsecloud_functionper archiviare gli input (file FASTQ), i genomi di riferimento, gli output (VCF e file di annotazione) e il codice della Cloud Function.
Google Cloud Function v2
- Funzione v2
genomic-dag-kickoffper attivare il DAG dell'analisi genomica. - Event-driven, basata sulla finalizzazione degli oggetti GCS. La funzione verifica la presenza di un file
ready.txte poi utilizza l'elenco delle posizioni dei bucket FASTQ contenuto in quel file per avviare il DAG che li elaborerà.
Istanza Google Compute Engine
grabbing-reference-genomeè un'istanza c4-standard-96 che scarica due genomi di riferimento felini da NCBI, li indicizza con GATK e poi carica i genomi di riferimento elaborati nel bucketgenomic_reference, dove saranno usati in seguito dal DAG.- Questa risorsa va eseguita una sola volta e si termina automaticamente al termine del lavoro.
Una volta che le risorse create da terraform apply sono attive e in funzione, per avviare l'analisi del genoma felino basta:
- caricare i FASTQ di BasePaws nel bucket
genomic_inputs_<random_id>; - creare un file
ready.txtcontenente gli URI GCS di quei FASTQ e caricarlo nello stesso bucket.
Il caricamento del file ready.txt attiverà la Cloud Function genomic-dag-kickoff, che avvierà l'esecuzione di due istanze del DAG su Cloud Composer, ciascuna delle quali orchestrerà le analisi secondaria e terziaria su una delle due versioni più recenti del genoma felino: Felis_catus_9.0 e Felis_catus_Fca126_mat1.0.
Il DAG esegue una complessa orchestrazione di strumenti bioinformatici che alla fine restituisce:
- i file VCF, ovvero Variant Call Files, cioè l'elenco di tutte le mutazioni individuate;
- i file di annotazione prodotti da SnpEff. SnpEff fornisce annotazioni in silico per ogni mutazione localizzata all'interno di un gene codificante per proteine, indicando la probabilità che tale mutazione produca un impatto funzionale 'Basso', 'Moderato' o 'Alto' sulla proteina codificata da quel gene.
A livello generale, il DAG è composto dai seguenti Task Group: i Task Group 'alignment' e 'variant calling' corrispondono all'Analisi Secondaria, mentre il Task Group 'annotation' rappresenta l'Analisi Terziaria.
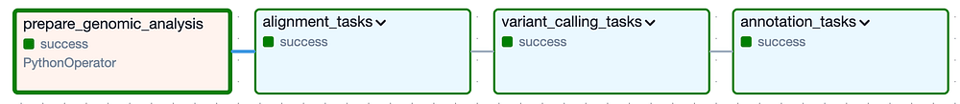
Il DAG di Cloud Composer per l'esecuzione delle analisi secondaria e terziaria su una specifica versione del genoma felino
Espandendo ciascun Task Group (qui sotto) emergono i numerosi task individuali necessari per eseguire questa pipeline di strumenti e poi verificare che siano stati creati i file di output attesi. Ho incluso una logica che salta interamente un Task Group se i suoi file di output sono già presenti nel bucket di output da un'esecuzione precedente andata a buon fine.
Senza Apache Airflow e la versione completamente gestita da GCP di questo strumento open-source, concatenare in pipeline questi tool e verificarne la corretta esecuzione (o quella precedente già riuscita) a ogni passaggio sarebbe molto più complesso e dispendioso in termini di tempo. E non si tratterebbe solo di orchestrare i passaggi, ma anche di recuperarne e archiviarne i log, visualizzare l'avanzamento della pipeline in tempo reale e segnalare eventuali problemi.
Composer rende semplice monitorare l'avanzamento, creare pipeline con percorsi logici di branching e fare in modo che gli errori vengano intercettati e i relativi log presentati in una UI facilmente leggibile. Per questi motivi e per molti altri — come la capacità di Composer di scalare in modo economicamente vantaggioso le risorse di calcolo in operazioni a scala enterprise — considero gli strumenti di workflow orchestration come Composer un tassello fondamentale per costruire pipeline di dati scalabili.
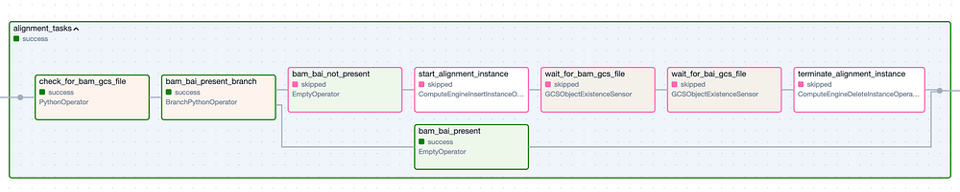
Il Task Group 'Alignment'. I passaggi con bordo verde rappresentano esecuzioni riuscite, quelli con bordo rosa sono stati saltati.
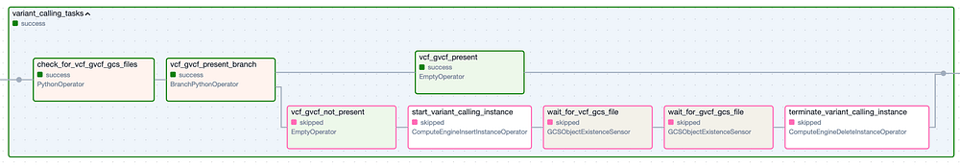
Il Task Group 'Variant Calling'. I passaggi con bordo verde rappresentano esecuzioni riuscite, quelli con bordo rosa sono stati saltati.
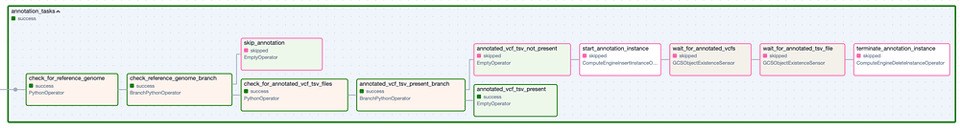
Il Task Group 'Annotation'. I passaggi con bordo verde rappresentano esecuzioni riuscite, quelli con bordo rosa sono stati saltati.
Una volta che tutti questi processi arrivano a completamento, vedrete VCF e file di annotazione depositarsi nel bucket GCS genomic-outputs-<random_id>. I VCF contengono l'elenco di tutte le mutazioni individuate, mentre i file di annotazione contengono i dettagli degli effetti predetti in silico da SnpEff sulle proteine codificate dai geni interessati da quelle mutazioni.
Ci interessa in particolare capire come, secondo SnpEff, i geni codificanti per proteine sono presumibilmente impattati.
È qui che ho iniziato davvero ad apprezzare il servizio di BasePaws. Nella genomica umana esiste una quantità sorprendente di fonti open-source di annotazioni delle varianti da cui attingere per ottenere una comprensione clinica approfondita di praticamente qualsiasi mutazione presente in un gene codificante per proteine. Nella genomica felina, invece, non sono riuscito a trovare alcun database di annotazione open-source, nemmeno uno per le mutazioni con esiti clinici chiari e conosciuti. Non avendo voglia di setacciare articoli accademici per costruirmi un database, mi è rimasto solo da implementare SnpEff nello step di annotazione del mio DAG di Cloud Composer, che ci fornirà almeno previsioni in silico degli effetti funzionali a cui appoggiarci. BasePaws deve aver investito moltissimo nello sviluppo interno di database di annotazione per poter offrire il tipo di analisi che ho visto nel report che mi hanno fornito.
Determinato comunque a trarre qualche utilità dalle previsioni di effetto di SnpEff, mi sono rivolto agli LLM, in particolare a Claude 3.5 Sonnet su AWS Bedrock.
Ho iniziato lanciando i seguenti comandi bash sul file di annotazione prodotto da SnpEff. Sono comandi semplici che filtrano i 13,5 milioni di mutazioni annotate riducendoli a sole 366 con maggiori probabilità di essere 'interessanti', ovvero quelle che:
- si trovano in una regione codificante per proteine;
- hanno maggiore probabilità di essere varianti reali piuttosto che chiamate errate;
- hanno un omologo umano identificato (un LLM ha più probabilità di comprenderne l'impatto potenziale se conosce la proteina umana equivalente);
- hanno un impatto previsto 'HIGH' sull'effetto funzionale di una proteina.
head -n1 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff.ann.tsv > 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact.ann.tsv
grep "\tHIGH\t.*protein_coding" 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff.ann.tsv >> 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact.ann.tsv
grep -v "frameshift" 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact.ann.tsv |
grep -v "intron" |
awk '$11 !~ /^ENSFCAG000/' > 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact_filtered.ann.tsv
cut -f 1,2,4,5,8-11,17,18,19,21 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact_filtered.ann.tsv
Ho incollato il contenuto di quel file filtrato, con le 366 varianti annotate ad impatto 'HIGH', in Claude, insieme al seguente prompt:
Analyze the following SnpEff genetic annotations for Felis catus 9.0. When referencing a gene, make sure to mention how many mutations were discovered in that gene and do not hallucinate mutations for genes that are not provided in the annotation file pasted below. Please provide:
1. A summary of the most significant findings
2. Highlight any mutations in genes associated with known health conditions, phenotypic traits, or important biological processes.
3. Explain the potential functional implications of the top 5–10 most interesting mutations, considering the gene's function and the type of mutation.
4. Identify any patterns, clusters of mutations, or impacted biological/functional pathways that might be noteworthy.
5. Suggest any potential health concerns or traits that might be affected based on these mutations, but clarify that this is speculative without clinical correlation.
Please use scientific terminology where appropriate, but also provide layman explanations to ensure understanding. If you need any clarification or additional information about the subject's phenotype or health history to improve your analysis, please ask.
The cat has an all white coat, is 11 years old, and has no known health issues other than feline tooth resorption. The cat has no known neurological or immune system disabilities (aside from the possible link between immune system function and feline tooth resorption).
Il risultato ottenuto è stato piuttosto interessante: Claude ipotizza una possibile predisposizione genetica al riassorbimento dentale felino sulla base di alcuni dei geni con mutazioni ad alto impatto. Trattandosi di un gatto dal mantello completamente bianco, è utile sapere che potrebbe essere anche più esposto al rischio di tumori a causa di una possibile compromissione di una proteina di riparazione del DNA: significa, in concreto, che dovrei limitare il suo tempo di esposizione alla luce solare diretta.
Gli altri risultati di Claude sono però molto speculativi: non ho infatti osservato nessuno dei gravi problemi riferiti a livello di funzione muscolare o metabolismo. Poiché non esistono dataset di annotazione ben organizzati e disponibili pubblicamente, la funzione dei geni felini è in larga parte inferita per omologia con i geni umani equivalenti. Mi aspetto quindi che gli LLM rispondano con inferenze ampie e sbilanciate sull'uomo, generando i risultati eccessivamente speculativi che Claude ha prodotto.
Thank you for providing this detailed genetic information about your 11-year-old white cat with feline tooth resorption. I'll analyze the high-impact mutations based on the SnpEff annotations you've shared. Let's break this down into the requested sections:
1. Summary of Most Significant Findings:
The genetic data reveals a substantial number of high-impact mutations across various genes. These mutations include stop-gained, start-lost, and stop-lost variants, which can significantly alter protein function. In total, there are over 200 high-impact mutations identified across numerous genes.
2. Mutations in Genes Associated with Known Conditions or Traits:
a) LRRC45 (1 mutation): Associated with ciliary function, which can affect various physiological processes.
b) PNKP (1 mutation): Involved in DNA repair, potentially impacting cancer susceptibility and neurological function.
c) ATP2A1 (1 mutation): Critical for muscle function, particularly in fast-twitch muscle fibers.
d) COQ7 (1 mutation): Important for coenzyme Q10 biosynthesis, affecting energy production in cells.
e) ABCC6 (1 mutation): Associated with connective tissue disorders in humans.
f) CRP (1 mutation): Involved in immune response and inflammation.
3. Potential Functional Implications of Top Mutations:
a) PNKP (c.1150C>T, p.Gln384*): This stop-gained mutation could lead to a truncated protein, potentially impairing DNA repair mechanisms. This might increase susceptibility to DNA damage and theoretically could affect neurological function or cancer risk.
b) ATP2A1 (c.3149A>G, p.Ter1050Trpext*?): This stop-lost mutation might result in an extended protein. ATP2A1 is crucial for muscle contraction, and alterations could potentially affect muscle function.
c) COQ7 (c.817T>A, p.Ter273Lysext*?): Another stop-lost mutation, this could lead to an extended protein. COQ7 is vital for coenzyme Q10 production, which is essential for cellular energy production. Alterations might affect overall energy metabolism.
d) ABCC6 (c.4246C>T, p.Arg1416*): This stop-gained mutation could result in a truncated protein. In humans, ABCC6 mutations are associated with pseudoxanthoma elasticum, a condition affecting connective tissue.
e) CRP (c.592T>C, p.Ter198Glnext*?): This stop-lost mutation in the C-reactive protein gene could affect inflammatory responses and immune function.
4. Patterns and Noteworthy Observations:
There appears to be a cluster of mutations affecting genes involved in muscle function, energy metabolism, and immune response. Several mutations are found in genes related to DNA repair and cellular maintenance processes.
5. Potential Health Concerns or Traits (Speculative):
Based on these mutations, there might be potential concerns related to:
\* Muscle function and metabolism (ATP2A1, COQ7)
\* Immune system regulation (CRP)
\* DNA repair and potential cancer susceptibility (PNKP)
\* Connective tissue health (ABCC6)
However, it's crucial to emphasize that the presence of these mutations doesn't necessarily mean the cat will develop related conditions. Many factors, including environmental influences and other genetic modifiers, play a role in gene expression and disease development.
It's interesting to note that despite these mutations, your cat has lived to 11 years with no major health issues reported aside from feline tooth resorption. This underscores the complex relationship between genotype and phenotype.
Given the cat's history of feline tooth resorption, the mutations in genes related to immune function (like CRP) and connective tissue (like ABCC6) might be of particular interest, as these systems are thought to play a role in this condition. However, a direct link cannot be established without further research.
A quel punto ho deciso di indagare quali geni sono noti per essere associati al mantello bianco nei gatti, per poi filtrare tutte le mutazioni annotate da SnpEff in quei geni indipendentemente dalla previsione di impatto basso/medio/alto. L'ho ottenuto filtrando il file di annotazione in cerca di mutazioni nei geni KIT e KITLG:
head -n1 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff.ann.tsv > 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_LOW_MODERATE_impact_white_coat.ann.tsv
grep -E "\t(LOW|MODERATE)\t.*protein_coding" 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff.ann.tsv | grep -E '\tKIT\t|\tKITLG\t' >> 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_LOW_MODERATE_impact_white_coat.ann.tsv
cut -f 1,2,4,5,8-11,17,18,19,21 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_LOW_MODERATE_impact_white_coat.ann.tsv
Riutilizzando lo stesso prompt, Claude ipotizza che le mutazioni missenso e quelle nei siti di splicing degli introni siano probabilmente responsabili del mantello bianco di Cloud:
Thank you for providing the genetic annotations for Felis catus 9.0. I'll analyze this data and provide the requested information. Let's break down the findings:
1. Summary of the most significant findings:
The annotations reveal multiple mutations in two important genes: KIT and KITLG. These genes are part of the KIT/KITLG signaling pathway, which plays crucial roles in various biological processes, including coat color development, hematopoiesis, and gametogenesis.
KIT gene: 9 mutations (7 synonymous variants, 2 splice region variants)
KITLG gene: 2 mutations (1 missense variant, 1 synonymous variant)
The most significant finding is the missense variant in the KITLG gene, which could potentially affect protein function.
2. Mutations associated with known health conditions, phenotypic traits, or important biological processes:
The KIT and KITLG genes are associated with coat color in cats, particularly white spotting patterns. The all-white coat of this cat might be related to these mutations, especially considering the number of variants in the KIT gene.
3. Potential functional implications of the top 5–10 most interesting mutations:
a) KITLG missense variant (c.731G>A, p.Ser244Asn): This mutation changes a serine to an asparagine at position 244 in the KITLG protein. This could potentially affect the protein's function, as it alters the amino acid sequence. KITLG is crucial for the survival and proliferation of melanocytes, which produce pigment. This mutation might contribute to the cat's all-white coat.
b) KIT splice region variants (c.2491–3C>T, c.1353–5T>C, c.1122–4_1122–3delGT): These mutations occur in splice regions, which are important for proper mRNA processing. Although they're classified as low impact, they could potentially affect the splicing of the KIT gene, leading to altered protein production or function. The KIT protein is a receptor tyrosine kinase involved in various developmental processes, including melanocyte development.
c) KIT synonymous variants: While synonymous variants don't change the amino acid sequence, they can still impact gene expression through various mechanisms such as altering mRNA stability or translation efficiency. The high number of synonymous variants in the KIT gene (7 in total) might collectively influence its expression or function.
4. Patterns, clusters of mutations, or impacted biological pathways:
The mutations are clustered in the KIT/KITLG signaling pathway, which is crucial for melanocyte development and function. This pathway is also involved in hematopoiesis (blood cell formation) and gametogenesis (production of reproductive cells). The concentration of mutations in these genes suggests a potential impact on pigmentation and possibly other related processes.
5. Potential health concerns or traits that might be affected (speculative):
a) Coat color: The all-white coat of the cat is likely related to these mutations, particularly in the KIT and KITLG genes.
b) Hearing: Some all-white cats with KIT mutations are prone to congenital deafness. While no specific deafness-associated mutations are present here, it might be worth monitoring the cat's hearing.
c) Immune function: The KIT/KITLG pathway plays a role in hematopoiesis, including the development of certain immune cells. While the cat has no known immune system disabilities, these mutations could potentially influence immune function subtly.
d) Tooth resorption: Although not directly linked to KIT/KITLG in current literature, the pathway's involvement in various developmental processes might indirectly contribute to dental issues. This is highly speculative and would require further research to establish any potential connection.
e) Fertility: Given the role of KIT/KITLG in gametogenesis, there could be potential effects on fertility. However, this is speculative and would only be relevant if the cat were to be bred.
It's important to note that while these genetic findings are interesting, their clinical significance can only be determined through correlation with the cat's actual health status and further studies. The cat's current good health at 11 years old suggests that many of these mutations may not have severe impacts.
Ciò che porto a casa da tutto questo è che è possibile ricavare risultati piuttosto interessanti dai dati genomici del proprio gatto, e che ottenere quei dati genomici diventa nettamente più realizzabile, affidabile e scalabile grazie ai servizi cloud che orchestrano le pipeline di dati, come Cloud Composer.
Restano però molte cose da fare sul fronte della ricerca finanziata pubblicamente. Inferire la funzionalità dei geni felini dalla loro omologia con quelli umani consente analisi significative solo entro certi limiti. Quando le istituzioni di ricerca veterinaria inizieranno a pubblicare database di annotazione genomica per gatti e cani, credo che vedremo davvero brillare le analisi dei dati genomici basate su LLM. Fino ad allora, resto colpito dal lavoro scientifico portato avanti dal team di Basepaws.
Ed eccoci qui! Abbiamo attraversato l'affascinante frontiera della genomica felina, dall'eccellente report di BasePaws fino alla nostra analisi DIY alimentata da servizi di cloud computing all'avanguardia e dall'AI generativa. Orchestrare una sinfonia di strumenti genomici può essere complicato come radunare un branco di gatti, ma con servizi come Cloud Composer diventa decisamente più semplice.
PerfectScale™ for Kubernetes
Ready to optimize?
Get your free Kubernetes savings analysis
Your cloud bill shouldn't be a mystery
Optimization, automation, expertise. In one platform.