A internet le encantan los gatos, así que supongo que Cloud me traerá más visitas que todos mis otros artículos juntos.
Cuando mi esposa me regaló las Navidades pasadas un kit de Basepaws Whole Genome Sequencing para nuestro adorado gato Cloud, quedé fascinado. No solo porque es alucinante que esta tecnología exista y sea comercialmente viable, sino porque, como bioinformático acostumbrado a analizar datos genómicos humanos, sé lo difícil que es descubrir y reportar hallazgos verdaderamente relevantes a partir de un genoma comparativamente menos estudiado como el de Felis catus.
Tenía curiosidad por ver hasta qué punto esta empresa podía ofrecer información interesante o accionable sobre mi mascota, y también qué tanto podría sacar yo mismo de los datos crudos de secuenciación al correr mis propios análisis. No es tarea sencilla: para completar análisis genómicos de manera costo-eficiente hay que encadenar muchas herramientas complejas en pipelines, y la ejecución de cada paso debe orquestarse para correr en máquinas de distintos tamaños y capacidades. ¿Y quién soy yo para rechazar semejante reto en honor a mi adorable felino?
Antes de explicar cómo aproveché la última versión de Cloud Composer (el servicio totalmente administrado de Apache Airflow en GCP) para simplificar la ejecución automatizada y la visualización de varios pasos complejos de análisis genómico, así como cómo usé Claude 3.5 Sonnet en AWS Bedrock para analizar los resultados, quiero destacar lo impresionado que quedé con el reporte en PDF generado por Basepaws.
Con sus 68 páginas, hay mucho más de lo que puedo cubrir en este artículo. En resumen, el reporte comunica con claridad sobre tu mascota:
- Datos de grupo de raza (más abajo se muestra una pequeña parte)
- Presencia o ausencia de marcadores genéticos de salud y trastornos genéticos
- Una gran cantidad de marcadores de rasgos interesantes
- Y, para mi sorpresa, un reporte de salud bucal basado en el microbioma oral
El reporte de salud bucal en particular me pareció genial: confirmó algunos problemas que ya conocía (por ejemplo, riesgo "Medio" de enfermedad periodontal) y también que las acciones tomadas para mitigar otros han dado resultado (por ejemplo, riesgo "Bajo" de reabsorción dental, un problema con probable origen parcialmente genético que ha tenido durante años y que mis veterinarios han manejado con éxito).
Ya es bastante difícil analizar el genoma del gato, que está relativamente poco anotado, pero secuenciar además el microbioma oral y entregar análisis significativos y accionables sobre eso es un verdadero logro. Felicitaciones al equipo de científicos que lo hizo posible.

He pensado en tener un Maine Coon como próximo gato. ¡No habría imaginado que ya tenía uno (más o menos)!
Dicho esto, exploremos qué se puede hacer con los datos crudos del genoma completo que entrega esta empresa junto con su reporte.
Más abajo encontrarás el enlace al repo asociado a este blog, que aprovecha la versión 3 recién lanzada de Cloud Composer y otros servicios de vanguardia de GCP —todo desplegado con el Terraform incluido— para automatizar, escalar y visualizar por completo la ejecución del complejo entramado de herramientas necesarias para realizar el análisis secundario y terciario de datos del genoma completo del gato.
https://github.com/doit-mattporter/genomics-workflow-orchestration
Empecemos esta charla sobre cómo correr análisis genómicos felinos en GCP con un repaso básico de lo que ejecutará este repo.
Para quienes no están familiarizados con la bioinformática: en términos generales, hay tres etapas principales en la pipeline que representan cómo se analizan los datos genómicos, sin importar el organismo. El análisis primario ya viene listo cuando recibes los datos de secuenciación de Basepaws, mientras que los análisis secundario y terciario se ejecutan con la ayuda del código que comparto aquí. Las tres etapas se describen a continuación:
- Análisis primario. Este paso ocurre en el centro de secuenciación de ADN y consiste en convertir los datos crudos de señal (datos de luz) de las máquinas de secuenciación en secuencias de pares de bases de ADN (cadenas de A/C/G/T). También incluye chequeos de QC, como descartar lecturas de baja confianza.
Por limitaciones técnicas, es imposible secuenciar una hebra de ADN de principio a fin en una sola lectura continua y enorme. Las hebras deben fragmentarse en millones de piezas cortas que recién entonces pueden secuenciarse. La salida del análisis primario serán, por lo tanto, millones —o incluso miles de millones— de fragmentos cortos de ADN, típicamente de 100 a 300 pares de bases (bps) de longitud cada uno. Estos datos crudos y fragmentados se entregan en formato FASTQ, que es básicamente texto plano con cadenas de valores ACGT para esos millones de secuencias cortas. Eso es lo que te envía BasePaws y con lo que comenzará a trabajar este blog y el repo asociado. 2. Análisis secundario. Esta etapa es como armar un rompecabezas gigante de tu ADN, donde la solución siempre difiere un poquito de lo que muestra la caja. Tiene 2 componentes principales:
a) "Sequence Alignment", o sea, ensamblado del rompecabezas de ADN: esos millones de fragmentos cortos del análisis primario se vuelven a unir —con muchísimo poder de cómputo— para reconstruir el genoma original e íntegro del gato del cual provienen.
b) "Variant Calling", o sea, encontrar las diferencias: una vez reensamblado el genoma de tu gato, se compara contra el genoma de "referencia" que la comunidad científica definió como estándar de oro de cómo luce un genoma típico de gato. Tu gato se desviará de ese estándar de "referencia" debido a su singularidad genética; este paso consiste en buscar esas diferencias, los lugares donde el ADN de tu gato difiere del ADN "típico". A esas diferencias se les llama "variantes" o "mutaciones". No todas las mutaciones detectadas durante el variant calling son reales: muchas son errores, por razones complejas. Este paso también incluye QC, para asegurarse de que las variantes son legítimas. 3. Análisis terciario. Suele ser la parte más complicada y crucial: descubrir qué significa realmente la lista única de diferencias de ADN encontradas en el análisis secundario. El análisis terciario representa la presentación de hallazgos tangibles y significativos. ¿Algunas de estas variantes tienen asociaciones conocidas con condiciones de salud? ¿Cómo responderá mi gato a distintas opciones farmacológicas según su lista de mutaciones? ¿Es esta mutación en particular la responsable del color de su pelaje? ¿Este conjunto de mutaciones indica que mi gato probablemente tiene ascendencia Maine Coon? El análisis terciario es la capacidad de responder preguntas de este tipo.
Ahora que ya tienes una idea suficiente del trabajo a realizar, ¡manos a la obra! Una vez que recibas los datos del genoma completo de tu gato desde BasePaws —o si solo quieres seguir el ejercicio y ver qué se puede hacer con Cloud Composer y Claude usando estos datos— puedes proceder así.
El README del repo describe que debes empezar provisionando la infraestructura cloud necesaria con una operación terraform apply. Ten en cuenta que algunos recursos, en particular Cloud Composer, correrán 24/7 hasta que los detengas, así que cuida tu gasto en cloud y asegúrate de terminar los recursos que no estés usando. Se desplegarán los siguientes recursos clave:
Entorno de Google Composer
- Entorno de Composer 3 con Apache Airflow 2
- Configura un DAG de Airflow para el procesamiento de datos genómicos felinos. Ten en cuenta que este DAG provisionará temporalmente instancias de Compute Engine c4-standard-96 y c4-standard-8 según lo requieran los distintos pasos de la pipeline.
Buckets de Google Cloud Storage
genomic_inputs,genomic_reference,genomic_outputsycloud_functionpara almacenar entradas (archivos FASTQ), genomas de referencia, salidas (VCFs y archivos de anotación) y el código de la Cloud Function
Google Cloud Function v2
- Función v2
genomic-dag-kickoffpara disparar el DAG de análisis genómico - Se activa por eventos de finalización de objetos en GCS. Esta función verifica si existe un archivo
ready.txty luego usa la lista de ubicaciones de buckets FASTQ que contiene para arrancar el DAG que procesará esos FASTQs.
Instancia de Google Compute Engine
grabbing-reference-genomees una instancia c4-standard-96 que descarga dos genomas de referencia felinos desde NCBI, los indexa con GATK y luego sube esos genomas de referencia procesados al bucketgenomic_referencepara que el DAG los use después.- Ten en cuenta que este recurso solo necesita correrse una vez y se autotermina al finalizar su trabajo.
Una vez que esos recursos estén corriendo a partir de terraform apply, basta con hacer lo siguiente para iniciar el análisis del genoma del gato:
- Sube los FASTQs de BasePaws al bucket
genomic_inputs_<random_id> - Crea un archivo
ready.txtque contenga las URIs de GCS de esos FASTQs y súbelo al mismo bucket.
La carga del archivo ready.txt disparará la Cloud Function genomic-dag-kickoff. Esta función iniciará la ejecución de dos instancias del DAG en Cloud Composer, cada una orquestando el análisis secundario y terciario contra una de las dos versiones más recientes del genoma del gato: Felis_catus_9.0 y Felis_catus_Fca126_mat1.0.
El DAG ejecuta una orquestación compleja de herramientas bioinformáticas que finalmente nos entrega:
- Archivos VCF, o Variant Call Files. Es la lista de todas las mutaciones descubiertas.
- Archivos de anotación generados por SnpEff. SnpEff aporta anotaciones in silico para cada mutación localizada dentro de un gen codificante de proteína, junto con la probabilidad de que esa mutación tenga un impacto funcional "Bajo", "Moderado" o "Alto" sobre la proteína codificada por ese gen.
A alto nivel, el DAG se compone de los siguientes Task Groups: los grupos "alignment" y "variant calling" corresponden al Análisis Secundario, mientras que el grupo "annotation" representa el Análisis Terciario:
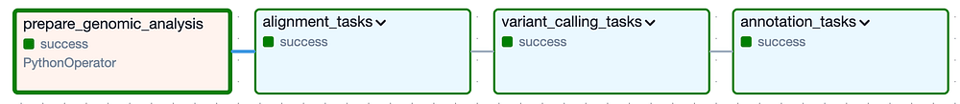
El DAG de Cloud Composer para ejecutar el análisis secundario y terciario sobre una versión específica del genoma del gato
Al expandir cada uno de los Task Groups (que se ven más abajo) aparecen las muchas tareas individuales necesarias para correr esta pipeline de herramientas y luego validar que se hayan creado los archivos de salida esperados. Vale la pena mencionar que incluí lógica para saltarse por completo cada Task Group si sus archivos de salida ya están presentes en el bucket de salida de una corrida exitosa anterior.
Sin la ayuda de Apache Airflow y de la versión totalmente administrada de esta herramienta open-source en GCP, encadenar estas herramientas y verificar su ejecución exitosa (o exitosa previa) en cada paso sería sustancialmente más complicado y lento de implementar. No solo sería un reto orquestar los pasos, sino también recolectar y guardar sus logs, visualizar el progreso de la pipeline en tiempo real y reportar cualquier incidencia.
Composer permite ver el progreso de manera fácil, crear pipelines con caminos de lógica ramificada y asegurarse de que los errores se capturen y sus logs se presenten en una UI fácil de leer. Por estas y muchas otras razones —como la capacidad de Composer para escalar recursos de cómputo de forma costo-eficiente cuando se usa en operaciones de escala empresarial— considero que las herramientas de orquestación de workflows como Composer son críticas para construir pipelines de datos escalables.
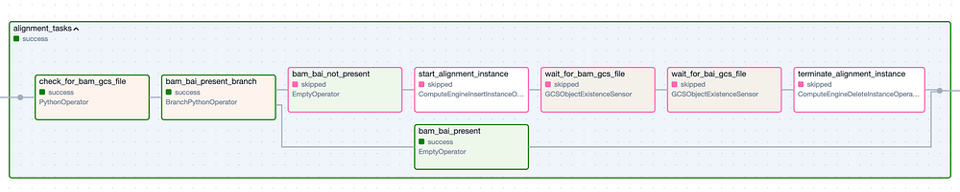
El Task Group "Alignment". Los pasos con borde verde representan ejecuciones exitosas, mientras que los de borde rosado fueron omitidos.
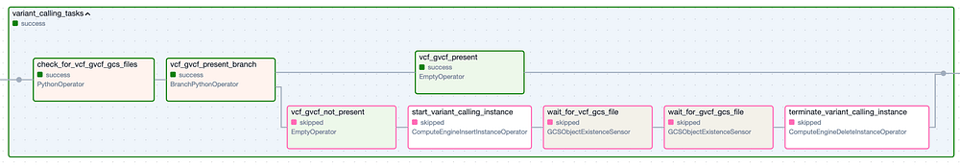
El Task Group "Variant Calling". Los pasos con borde verde representan ejecuciones exitosas, mientras que los de borde rosado fueron omitidos.
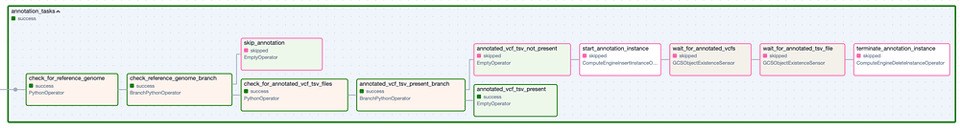
El Task Group "Annotation". Los pasos con borde verde representan ejecuciones exitosas, mientras que los de borde rosado fueron omitidos.
Cuando todo esto termine de correr, verás los VCFs y los archivos de anotación volcados en el bucket de GCS genomic-outputs-<random_id>. Los VCFs contienen la lista de todas las mutaciones descubiertas, mientras que los archivos de anotación incluyen los detalles de los efectos predichos in silico por SnpEff sobre las proteínas codificadas por los genes.
Lo que más nos interesa es cómo SnpEff cree que se verán afectados los genes codificantes de proteína.
Aquí es donde mi aprecio por el servicio de BasePaws empezó a crecer en serio. En la genómica humana existe una asombrosa cantidad de fuentes open-source de anotación de variantes a las que puedes recurrir para tener un entendimiento clínico profundo de prácticamente cualquier mutación que ocurra dentro de un gen codificante. En cambio, en el ámbito de la genómica felina no encontré ninguna base de datos de anotación open-source, ni siquiera una para mutaciones con desenlaces clínicos claros y conocidos. Como no tenía muchas ganas de raspar artículos académicos para armar mi propia base de datos, solo me quedó implementar SnpEff en el paso de anotación del DAG en Cloud Composer, lo que al menos nos da predicciones in silico de efecto funcional como apoyo. BasePaws debe haber invertido muchísimo esfuerzo en construir bases de datos de anotación internas para poder ofrecer el tipo de análisis que vi en el reporte que me entregaron.
En fin, decidido a sacarle algo de provecho a las predicciones de efecto de SnpEff, recurrí a los LLMs, específicamente a Claude 3.5 Sonnet corriendo en AWS Bedrock.
Empecé corriendo los siguientes comandos de bash sobre el archivo de anotación que produce SnpEff. Estos comandos sencillos filtran los 13.5 millones de mutaciones anotadas hasta dejar solo 366 que tienen más probabilidades de ser "interesantes": las que:
- Están dentro de una región codificante de proteína
- Tienen mayor probabilidad de ser variantes legítimas y no un variant call erróneo
- Tienen un homólogo humano identificado (un LLM va a entender mejor el posible impacto si conoce su equivalente humano), y
- Tienen un impacto funcional predicho como "HIGH" sobre la proteína
head -n1 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff.ann.tsv > 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact.ann.tsv
grep "\tHIGH\t.*protein_coding" 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff.ann.tsv >> 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact.ann.tsv
grep -v "frameshift" 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact.ann.tsv |
grep -v "intron" |
awk '$11 !~ /^ENSFCAG000/' > 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact_filtered.ann.tsv
cut -f 1,2,4,5,8-11,17,18,19,21 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact_filtered.ann.tsv
Pegué el contenido de ese archivo filtrado con 366 variantes anotadas de impacto "HIGH" en Claude, junto con el siguiente prompt:
Analyze the following SnpEff genetic annotations for Felis catus 9.0. When referencing a gene, make sure to mention how many mutations were discovered in that gene and do not hallucinate mutations for genes that are not provided in the annotation file pasted below. Please provide:
1. A summary of the most significant findings
2. Highlight any mutations in genes associated with known health conditions, phenotypic traits, or important biological processes.
3. Explain the potential functional implications of the top 5–10 most interesting mutations, considering the gene's function and the type of mutation.
4. Identify any patterns, clusters of mutations, or impacted biological/functional pathways that might be noteworthy.
5. Suggest any potential health concerns or traits that might be affected based on these mutations, but clarify that this is speculative without clinical correlation.
Please use scientific terminology where appropriate, but also provide layman explanations to ensure understanding. If you need any clarification or additional information about the subject's phenotype or health history to improve your analysis, please ask.
The cat has an all white coat, is 11 years old, and has no known health issues other than feline tooth resorption. The cat has no known neurological or immune system disabilities (aside from the possible link between immune system function and feline tooth resorption).
Esto arrojó los resultados que ves a continuación, que me parecieron bastante interesantes, ya que Claude sí sugiere una posible predisposición genética a la reabsorción dental felina basándose en algunos de los genes con mutaciones de alto impacto. Con un pelaje totalmente blanco, la posibilidad de que también esté en mayor riesgo de cáncer por la posible pérdida de funcionalidad de una proteína de reparación del ADN es algo bueno de tener presente, ya que implica que debería limitar su exposición directa al sol.
Los demás hallazgos de Claude son, sin embargo, bastante especulativos, ya que no he observado ninguno de los problemas serios reportados sobre función muscular o metabolismo. Como no existen datasets de anotación bien organizados y públicamente disponibles, la función de los genes felinos se infiere en gran medida por homología con sus equivalentes humanos. Por eso espero que los LLMs respondan con inferencias amplias y centradas en el ser humano, lo que lleva a los resultados excesivamente especulativos que produjo Claude.
Thank you for providing this detailed genetic information about your 11-year-old white cat with feline tooth resorption. I'll analyze the high-impact mutations based on the SnpEff annotations you've shared. Let's break this down into the requested sections:
1. Summary of Most Significant Findings:
The genetic data reveals a substantial number of high-impact mutations across various genes. These mutations include stop-gained, start-lost, and stop-lost variants, which can significantly alter protein function. In total, there are over 200 high-impact mutations identified across numerous genes.
2. Mutations in Genes Associated with Known Conditions or Traits:
a) LRRC45 (1 mutation): Associated with ciliary function, which can affect various physiological processes.
b) PNKP (1 mutation): Involved in DNA repair, potentially impacting cancer susceptibility and neurological function.
c) ATP2A1 (1 mutation): Critical for muscle function, particularly in fast-twitch muscle fibers.
d) COQ7 (1 mutation): Important for coenzyme Q10 biosynthesis, affecting energy production in cells.
e) ABCC6 (1 mutation): Associated with connective tissue disorders in humans.
f) CRP (1 mutation): Involved in immune response and inflammation.
3. Potential Functional Implications of Top Mutations:
a) PNKP (c.1150C>T, p.Gln384*): This stop-gained mutation could lead to a truncated protein, potentially impairing DNA repair mechanisms. This might increase susceptibility to DNA damage and theoretically could affect neurological function or cancer risk.
b) ATP2A1 (c.3149A>G, p.Ter1050Trpext*?): This stop-lost mutation might result in an extended protein. ATP2A1 is crucial for muscle contraction, and alterations could potentially affect muscle function.
c) COQ7 (c.817T>A, p.Ter273Lysext*?): Another stop-lost mutation, this could lead to an extended protein. COQ7 is vital for coenzyme Q10 production, which is essential for cellular energy production. Alterations might affect overall energy metabolism.
d) ABCC6 (c.4246C>T, p.Arg1416*): This stop-gained mutation could result in a truncated protein. In humans, ABCC6 mutations are associated with pseudoxanthoma elasticum, a condition affecting connective tissue.
e) CRP (c.592T>C, p.Ter198Glnext*?): This stop-lost mutation in the C-reactive protein gene could affect inflammatory responses and immune function.
4. Patterns and Noteworthy Observations:
There appears to be a cluster of mutations affecting genes involved in muscle function, energy metabolism, and immune response. Several mutations are found in genes related to DNA repair and cellular maintenance processes.
5. Potential Health Concerns or Traits (Speculative):
Based on these mutations, there might be potential concerns related to:
\* Muscle function and metabolism (ATP2A1, COQ7)
\* Immune system regulation (CRP)
\* DNA repair and potential cancer susceptibility (PNKP)
\* Connective tissue health (ABCC6)
However, it's crucial to emphasize that the presence of these mutations doesn't necessarily mean the cat will develop related conditions. Many factors, including environmental influences and other genetic modifiers, play a role in gene expression and disease development.
It's interesting to note that despite these mutations, your cat has lived to 11 years with no major health issues reported aside from feline tooth resorption. This underscores the complex relationship between genotype and phenotype.
Given the cat's history of feline tooth resorption, the mutations in genes related to immune function (like CRP) and connective tissue (like ABCC6) might be of particular interest, as these systems are thought to play a role in this condition. However, a direct link cannot be established without further research.
Después decidí investigar qué genes se conocen como asociados al color de pelaje blanco en los gatos, y luego me puse a filtrar todas las mutaciones anotadas por SnpEff dentro de esos genes, sin importar si la predicción de impacto era baja, media o alta. Esto se logró filtrando el archivo de anotación por mutaciones en los genes KIT y KITLG:
head -n1 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff.ann.tsv > 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_LOW_MODERATE_impact_white_coat.ann.tsv
grep -E "\t(LOW|MODERATE)\t.*protein_coding" 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff.ann.tsv | grep -E '\tKIT\t|\tKITLG\t' >> 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_LOW_MODERATE_impact_white_coat.ann.tsv
cut -f 1,2,4,5,8-11,17,18,19,21 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_LOW_MODERATE_impact_white_coat.ann.tsv
Reusando el mismo prompt, Claude conjetura que las mutaciones missense y de sitio de splicing en intrones son probablemente las responsables del color de pelaje blanco de Cloud:
Thank you for providing the genetic annotations for Felis catus 9.0. I'll analyze this data and provide the requested information. Let's break down the findings:
1. Summary of the most significant findings:
The annotations reveal multiple mutations in two important genes: KIT and KITLG. These genes are part of the KIT/KITLG signaling pathway, which plays crucial roles in various biological processes, including coat color development, hematopoiesis, and gametogenesis.
KIT gene: 9 mutations (7 synonymous variants, 2 splice region variants)
KITLG gene: 2 mutations (1 missense variant, 1 synonymous variant)
The most significant finding is the missense variant in the KITLG gene, which could potentially affect protein function.
2. Mutations associated with known health conditions, phenotypic traits, or important biological processes:
The KIT and KITLG genes are associated with coat color in cats, particularly white spotting patterns. The all-white coat of this cat might be related to these mutations, especially considering the number of variants in the KIT gene.
3. Potential functional implications of the top 5–10 most interesting mutations:
a) KITLG missense variant (c.731G>A, p.Ser244Asn): This mutation changes a serine to an asparagine at position 244 in the KITLG protein. This could potentially affect the protein's function, as it alters the amino acid sequence. KITLG is crucial for the survival and proliferation of melanocytes, which produce pigment. This mutation might contribute to the cat's all-white coat.
b) KIT splice region variants (c.2491–3C>T, c.1353–5T>C, c.1122–4_1122–3delGT): These mutations occur in splice regions, which are important for proper mRNA processing. Although they're classified as low impact, they could potentially affect the splicing of the KIT gene, leading to altered protein production or function. The KIT protein is a receptor tyrosine kinase involved in various developmental processes, including melanocyte development.
c) KIT synonymous variants: While synonymous variants don't change the amino acid sequence, they can still impact gene expression through various mechanisms such as altering mRNA stability or translation efficiency. The high number of synonymous variants in the KIT gene (7 in total) might collectively influence its expression or function.
4. Patterns, clusters of mutations, or impacted biological pathways:
The mutations are clustered in the KIT/KITLG signaling pathway, which is crucial for melanocyte development and function. This pathway is also involved in hematopoiesis (blood cell formation) and gametogenesis (production of reproductive cells). The concentration of mutations in these genes suggests a potential impact on pigmentation and possibly other related processes.
5. Potential health concerns or traits that might be affected (speculative):
a) Coat color: The all-white coat of the cat is likely related to these mutations, particularly in the KIT and KITLG genes.
b) Hearing: Some all-white cats with KIT mutations are prone to congenital deafness. While no specific deafness-associated mutations are present here, it might be worth monitoring the cat's hearing.
c) Immune function: The KIT/KITLG pathway plays a role in hematopoiesis, including the development of certain immune cells. While the cat has no known immune system disabilities, these mutations could potentially influence immune function subtly.
d) Tooth resorption: Although not directly linked to KIT/KITLG in current literature, the pathway's involvement in various developmental processes might indirectly contribute to dental issues. This is highly speculative and would require further research to establish any potential connection.
e) Fertility: Given the role of KIT/KITLG in gametogenesis, there could be potential effects on fertility. However, this is speculative and would only be relevant if the cat were to be bred.
It's important to note that while these genetic findings are interesting, their clinical significance can only be determined through correlation with the cat's actual health status and further studies. The cat's current good health at 11 years old suggests that many of these mutations may not have severe impacts.
Mi conclusión de todo esto es que sí es posible obtener hallazgos bastante interesantes a partir de los datos genómicos de tu gato, y que conseguirlos se vuelve mucho más factible, confiable y escalable con la ayuda de servicios cloud que orquestan pipelines de datos, como Cloud Composer.
Sin embargo, queda mucho por hacer del lado de la investigación financiada con fondos públicos. Inferir la funcionalidad de los genes felinos a partir de su homología con los humanos solo permitirá hacer análisis significativos hasta cierto punto. Cuando las instituciones de investigación veterinaria empiecen a publicar bases de datos de anotación genómica para gatos y perros, creo que veremos brillar de verdad a los análisis genómicos potenciados por LLMs. Hasta entonces, me quedo impresionado con el trabajo científico que ha hecho el equipo de Basepaws.
¡Y ahí lo tienes! Hemos recorrido la fascinante frontera de la genómica felina, desde el impresionante reporte de BasePaws hasta nuestro propio análisis DIY potenciado por servicios de cloud computing de última generación e IA generativa. Aunque orquestar una sinfonía de herramientas genómicas puede ser tan complicado como arrear gatos, servicios como Cloud Composer lo hacen mucho más sencillo.
PerfectScale™ for Kubernetes
Ready to optimize?
Get your free Kubernetes savings analysis
Your cloud bill shouldn't be a mystery
Optimization, automation, expertise. In one platform.