Das Internet liebt Katzen – und ich gehe davon aus, dass Cloud mir mehr Klicks beschert als alle meine bisherigen Blogposts zusammen.
Als meine Frau mir letzte Weihnachten ein Basepaws Whole Genome Sequencing-Kit für unseren geliebten Kater Cloud schenkte, war ich ziemlich begeistert. Nicht nur, weil es schlicht cool ist, dass es diese Technologie überhaupt gibt und sie kommerziell verfügbar ist, sondern auch, weil ich als Bioinformatiker so sehr daran gewöhnt bin, menschliche Genomdaten zu analysieren, dass ich gut nachvollziehen kann, wie schwierig es ist, aus einem vergleichsweise wenig erforschten Genom wie dem von Felis catus wirklich aussagekräftige Erkenntnisse zu gewinnen und aufzubereiten.
Mich interessierte, wie viel dieses Unternehmen tatsächlich an interessanten oder umsetzbaren Informationen über mein Haustier liefern kann – und wie viel ich selbst aus den Rohdaten der Sequenzierung herausholen würde. Keine triviale Aufgabe: Damit genomische Analysen kosteneffizient laufen, müssen viele komplexe Tools zu einer Pipeline verkettet und auf Maschinen unterschiedlicher Größe und Leistung orchestriert werden. Wer wäre ich, mich dieser Herausforderung für meinen kleinen Stubentiger zu verweigern?
Bevor ich darauf eingehe, wie ich mit der neuesten Version von Cloud Composer (dem vollständig verwalteten Apache-Airflow-Service von GCP) die automatisierte Ausführung und Visualisierung mehrerer komplexer Genomanalyseschritte vereinfacht und mit Claude 3.5 Sonnet auf AWS Bedrock die Ergebnisse ausgewertet habe, möchte ich kurz hervorheben, wie beeindruckt ich vom PDF-Report von Basepaws war.
Mit 68 Seiten enthält er weit mehr, als ich in diesem Artikel abdecken kann. Kurz zusammengefasst liefert der Report für Ihr Haustier:
- Daten zur Rassengruppe (ein Beispiel sehen Sie unten)
- Vorhandensein bzw. Fehlen genetischer Gesundheitsmarker und Erbkrankheiten
- Eine Vielzahl interessanter Merkmalsmarker
- Und – zu meiner Überraschung – einen Bericht zur Mundgesundheit auf Basis des oralen Mikrobioms
Gerade der Mundgesundheitsbericht hatte es in sich: Er bestätigte einige bereits bekannte Probleme (z. B. "mittleres" Risiko für Parodontalerkrankungen) und zeigte, dass Maßnahmen gegen andere Probleme erfolgreich gewirkt haben (z. B. "niedriges" Risiko für Zahnresorption – ein vermutlich teils genetisch bedingtes Problem, das er seit Jahren hat und das meine Tierärzte erfolgreich behandelt haben).
Das ohnehin schwach annotierte Katzengenom zu analysieren ist schon anspruchsvoll genug. Zusätzlich noch das orale Mikrobiom zu sequenzieren und daraus aussagekräftige, umsetzbare Analysen abzuleiten, ist eine echte Leistung. Hut ab vor dem Wissenschaftlerteam, das das auf die Beine gestellt hat.

Ich hatte überlegt, mir als nächste Katze eine Maine Coon zuzulegen – und hätte nie gedacht, dass ich (gewissermaßen) schon eine habe!
Schauen wir uns nun an, was Sie mit den rohen Whole-Genome-Daten anstellen können, die das Unternehmen Ihnen zusammen mit dem Report bereitstellt.
Unten verlinkt finden Sie ein Repo zu diesem Blogpost, das die kürzlich veröffentlichte v3 von Cloud Composer und einige weitere topaktuelle GCP-Services nutzt – komplett über das mitgelieferte Terraform bereitgestellt. Damit lässt sich die Ausführung des komplexen Tool-Geflechts für die sekundäre und tertiäre Analyse von Katzen-Whole-Genome-Daten vollständig automatisieren, skalieren und visualisieren.
https://github.com/doit-mattporter/genomics-workflow-orchestration
Bevor wir uns ansehen, wie genomische Analysen für Katzen in GCP umgesetzt werden, lohnt ein grundlegendes Verständnis dessen, was dieses Repo eigentlich ausführt.
Für alle, die mit Bioinformatik nicht vertraut sind: Grundsätzlich gibt es drei zentrale Pipeline-Stufen, die abbilden, wie Genomdaten analysiert werden – unabhängig vom Organismus. Die primäre Analyse ist bereits abgeschlossen, wenn Sie Ihre Sequenzierungsdaten von Basepaws erhalten, während die sekundäre und tertiäre Analyse mithilfe der von mir bereitgestellten Codebasis laufen. Die drei Stufen im Überblick:
- Primäre Analyse. Dieser Schritt findet im Sequenzierlabor statt und umfasst die Umwandlung der rohen Signaldaten (Lichtdaten) der Sequenziermaschinen in DNA-Basenpaarsequenzen (A/C/G/T-Sequenzen). Hinzu kommen QC-Prüfungen, etwa das Aussortieren von Sequenzlesungen mit geringer Konfidenz.
Aus technischen Gründen lässt sich ein DNA-Strang nicht in einer einzigen, langen, durchgehenden Lesung von Anfang bis Ende sequenzieren. Stattdessen werden DNA-Stränge in Millionen kurzer Stücke zerlegt, die sich erst dann sequenzieren lassen. Das Ergebnis der primären Analyse sind also Millionen – wenn nicht Milliarden – kurzer DNA-Fragmente, typischerweise 100 bis 300 Basenpaare (bp) lang. Diese rohen, fragmentierten Sequenzierungsdaten erhalten Sie im FASTQ-Format, im Wesentlichen einem reinen Textformat, das aus ACGT-Basenpaarketten für diese Millionen kurzer Sequenzen besteht. Genau das schickt Ihnen BasePaws – und damit setzt dieser Blogpost bzw. das zugehörige Repo an. 2. Sekundäre Analyse. Diese Stufe ist wie das Zusammensetzen eines riesigen DNA-Puzzles, bei dem die Lösung immer ein wenig anders aussieht als das Bild auf der Schachtel. Sie besteht aus zwei Hauptkomponenten:
a) "Sequence Alignment" alias DNA-Puzzle-Zusammenbau: Die Millionen kurzer DNA-Fragmente aus der primären Analyse werden – mit erheblichem Rechenaufwand – wieder zusammengesetzt, sodass das ursprüngliche, vollständige Katzengenom rekonstruiert wird, aus dem die Fragmente stammen.
b) "Variant Calling" alias "Finde die Unterschiede": Sobald das Genom Ihrer Katze rekonstruiert ist, wird es mit dem "Referenzgenom" verglichen, das die wissenschaftliche Gemeinschaft als Goldstandard für ein typisches Katzengenom etabliert hat. Aufgrund ihrer genetischen Einzigartigkeit weicht Ihre Katze von dieser Referenz ab; in diesem Schritt geht es darum, genau diese Unterschiede zu finden – Stellen, an denen sich die DNA Ihrer Katze von der "typischen" Katzen-DNA unterscheidet. Diese Unterschiede heißen "Varianten" oder "Mutationen". Nicht alle beim Variant Calling entdeckten Mutationen sind echt – aus verschiedenen, technisch komplexen Gründen sind viele Varianten Artefakte. Auch dieser Schritt umfasst QC, um sicherzustellen, dass die Varianten tatsächlich real sind. 3. Tertiäre Analyse. Oft der kniffligste und wichtigste Teil: Hier geht es darum herauszufinden, was die einzigartige Liste an DNA-Unterschieden aus der sekundären Analyse tatsächlich bedeutet. Die tertiäre Analyse liefert die greifbaren, aussagekräftigen Erkenntnisse. Sind einige dieser Varianten mit bekannten Gesundheitsproblemen assoziiert? Wie wird meine Katze auf verschiedene pharmazeutische Wirkstoffe reagieren – ausgehend von ihrer Mutationsliste? Ist eine bestimmte Mutation für ihre Fellfarbe verantwortlich? Deutet diese Mutationskombination darauf hin, dass meine Katze Maine-Coon-Vorfahren hat? Genau solche Fragen lassen sich mit der tertiären Analyse beantworten.
Jetzt, da Sie eine ausreichend gute Vorstellung von der anstehenden Arbeit haben, geht es los! Sobald Sie die Whole-Genome-Daten Ihrer Katze von BasePaws erhalten haben – oder wenn Sie einfach mitverfolgen wollen, was sich mit Cloud Composer und Claude aus solchen Daten herausholen lässt – können Sie wie folgt vorgehen.
In der README des Repos ist beschrieben, dass Sie zunächst die nötige Cloud-Infrastruktur per terraform apply bereitstellen sollten. Achten Sie darauf: Einige Ressourcen, insbesondere Cloud Composer, laufen rund um die Uhr, bis Sie sie beenden. Behalten Sie also Ihre Cloud-Kosten im Blick und beenden Sie ungenutzte Ressourcen rechtzeitig. Die folgenden Schlüsselressourcen werden eingerichtet:
Google Composer Environment
- Composer-3-Umgebung mit Apache Airflow 2
- Konfiguriert einen Airflow-DAG für die Verarbeitung genomischer Katzendaten. Beachten Sie, dass dieser DAG bei Bedarf vorübergehend Compute-Engine-Instanzen vom Typ c4-standard-96 und c4-standard-8 für einzelne Pipeline-Schritte bereitstellt.
Google Cloud Storage Buckets
genomic_inputs,genomic_reference,genomic_outputsundcloud_function– zur Ablage von Inputs (FASTQ-Dateien), Referenzgenomen, Outputs (VCFs und Annotationsdateien) sowie Cloud-Function-Code
Google Cloud Function v2
genomic-dag-kickoff-Funktion v2 zum Auslösen des Genomik-Analytics-DAGs- Event-getrieben durch die Finalisierung von GCS-Objekten. Die Funktion prüft auf eine
ready.txt-Datei und nutzt dann die darin aufgeführten FASTQ-Bucket-Speicherorte, um den DAG zur Verarbeitung dieser FASTQs anzustoßen.
Google Compute Engine-Instanz
grabbing-reference-genomeist eine c4-standard-96-Instanz, die zwei Katzen-Referenzgenome von NCBI herunterlädt, mit GATK indiziert und die verarbeiteten Referenzgenome anschließend in dengenomic_reference-Bucket hochlädt, damit der DAG sie später nutzen kann.- Diese Ressource muss nur einmal laufen und beendet sich nach Abschluss ihrer Arbeit selbst.
Sobald diese Ressourcen über terraform apply eingerichtet sind und laufen, müssen Sie nur noch Folgendes tun, um die Genomanalyse für Ihre Katze anzustoßen:
- Laden Sie die BasePaws-FASTQs in den Bucket
genomic_inputs_<random_id>hoch - Erstellen Sie eine
ready.txt-Datei mit den GCS-URIs dieser FASTQs und laden Sie sie in denselben Bucket hoch.
Das Hochladen der ready.txt-Datei löst die Cloud Function genomic-dag-kickoff aus. Diese startet auf Cloud Composer zwei DAG-Instanzen, die jeweils die sekundäre und tertiäre Analyse gegen eine der beiden aktuellsten Versionen des Katzengenoms orchestrieren: Felis_catus_9.0 und Felis_catus_Fca126_mat1.0.
Der DAG führt eine komplexe Orchestrierung von Bioinformatik-Tools aus, die uns am Ende Folgendes liefert:
- VCF-Dateien (Variant Call Files). Eine Liste aller entdeckten Mutationen.
- Von SnpEff erzeugte Annotationsdateien. SnpEff liefert in-silico-Annotationen für jede Mutation in einem proteincodierenden Gen samt Wahrscheinlichkeit, dass diese Mutation eine "niedrige", "moderate" oder "hohe" funktionale Auswirkung auf das vom Gen codierte Protein hat.
Auf hoher Ebene besteht der DAG aus den folgenden Task Groups: "alignment" und "variant calling" entsprechen der sekundären Analyse, "annotation" steht für die tertiäre Analyse:
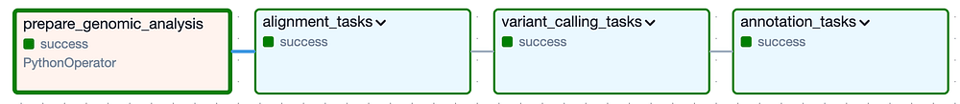
Cloud Composers DAG zur Ausführung der sekundären und tertiären Analyse für eine bestimmte Version des Katzengenoms
Klappt man die einzelnen Task Groups (siehe unten) auf, werden die vielen einzelnen Tasks sichtbar, die nötig sind, um die Tool-Pipeline auszuführen und anschließend zu prüfen, ob die erwarteten Output-Dateien erzeugt wurden. Ich habe übrigens eine Logik eingebaut, die eine Task Group komplett überspringt, wenn ihre Output-Dateien aus einem zuvor erfolgreichen Lauf bereits im Output-Bucket liegen.
Ohne Apache Airflow und GCPs vollständig verwaltete Variante dieses Open-Source-Tools wäre die Verkettung dieser Tools sowie die Verifizierung ihrer erfolgreichen (oder zuvor erfolgreichen) Ausführung in jedem Schritt deutlich aufwendiger und zeitintensiver. Nicht nur die Orchestrierung wäre eine Herausforderung, sondern auch das Abrufen und Speichern der Logs, die Echtzeit-Visualisierung des Pipeline-Fortschritts und das Reporting etwaiger Probleme.
Composer macht es einfach, den Fortschritt zu verfolgen, Pipelines mit verzweigter Ausführungslogik aufzubauen und sicherzustellen, dass Fehler erfasst und ihre Logs in einer übersichtlichen UI dargestellt werden. Aus diesen und vielen weiteren Gründen – etwa Composers Fähigkeit, Compute-Ressourcen im Enterprise-Maßstab kosteneffizient hoch- und herunterzuskalieren – halte ich Workflow-Orchestrierungstools wie Composer für unverzichtbar, wenn man skalierbare Daten-Pipelines bauen will.
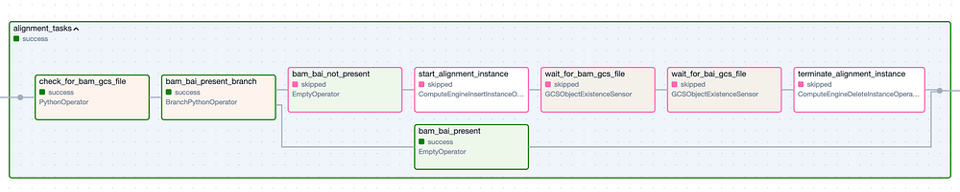
Die Task Group "Alignment". Schritte mit grünem Rand stehen für erfolgreiche Ausführungen, Schritte mit rosa Rand wurden übersprungen.
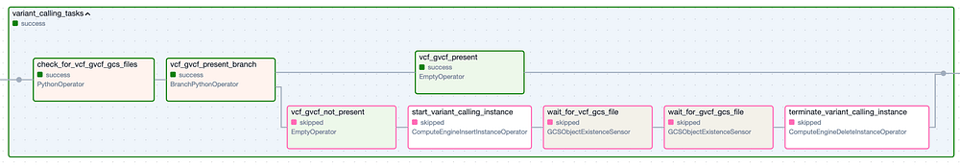
Die Task Group "Variant Calling". Schritte mit grünem Rand stehen für erfolgreiche Ausführungen, Schritte mit rosa Rand wurden übersprungen.
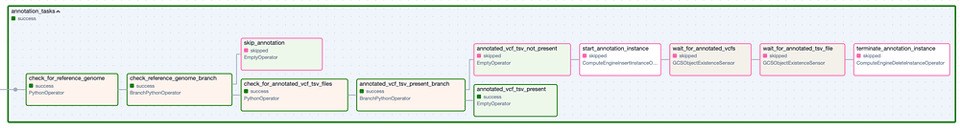
Die Task Group "Annotation". Schritte mit grünem Rand stehen für erfolgreiche Ausführungen, Schritte mit rosa Rand wurden übersprungen.
Sobald alles vollständig durchgelaufen ist, landen die VCFs und Annotationsdateien im GCS-Bucket genomic-outputs-<random_id>. Die VCFs enthalten eine Liste sämtlicher entdeckter Mutationen, die Annotationsdateien die Details zu SnpEffs in-silico-Vorhersagen über die Auswirkungen dieser Mutationen auf die von Genen codierten Proteine.
Uns interessiert insbesondere, wie SnpEff die wahrscheinliche Beeinträchtigung proteincodierender Gene einschätzt.
An dieser Stelle wurde mir die Leistung des BasePaws-Service so richtig bewusst. In der Humangenomik gibt es eine erstaunliche Fülle an Open-Source-Datenquellen für Variantenannotationen, aus denen man schöpfen kann, um ein tiefes klinisches Verständnis nahezu jeder Mutation in einem proteincodierenden Gen zu gewinnen. In der Katzengenomik dagegen konnte ich keine einzige Open-Source-Annotationsdatenbank finden – nicht einmal eine für Mutationen mit klaren, bekannten klinischen Folgen. Da ich keine Lust hatte, Fachpublikationen zu scrapen, um eine eigene Datenbank aufzubauen, blieb mir nur, SnpEff in den Annotationsschritt meines Cloud-Composer-DAGs einzubinden – damit haben wir zumindest in-silico-Vorhersagen funktionaler Effekte als Grundlage. BasePaws muss erheblich Aufwand in den internen Aufbau von Annotationsdatenbanken gesteckt haben, um die Art von Analysen liefern zu können, die ich in dem bereitgestellten Report gesehen habe.
Fest entschlossen, etwas aus den Effektvorhersagen von SnpEff herauszuholen, wandte ich mich also LLMs zu – konkret Claude 3.5 Sonnet auf AWS Bedrock.
Zunächst habe ich die folgenden Bash-Befehle auf die von SnpEff erzeugte Annotationsdatei angewendet. Diese einfachen Befehle filtern die 13,5 Millionen annotierten Mutationen auf nur 366 herunter, die mit höherer Wahrscheinlichkeit "interessant" sind – nämlich solche, die:
- in einer proteincodierenden Region liegen
- mit höherer Wahrscheinlichkeit echte Varianten und keine fehlerhaften Variant Calls sind
- ein identifiziertes menschliches Homolog besitzen (ein LLM kann die mögliche Auswirkung besser einschätzen, wenn es das menschliche Pendant kennt) und
- laut Vorhersage eine "HIGH"-Auswirkung auf die Funktion eines Proteins haben
head -n1 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff.ann.tsv > 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact.ann.tsv
grep "\tHIGH\t.*protein_coding" 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff.ann.tsv >> 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact.ann.tsv
grep -v "frameshift" 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact.ann.tsv |
grep -v "intron" |
awk '$11 !~ /^ENSFCAG000/' > 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact_filtered.ann.tsv
cut -f 1,2,4,5,8-11,17,18,19,21 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_HIGH_impact_filtered.ann.tsv
Den Inhalt dieser gefilterten Datei mit 366 als "HIGH" annotierten Varianten habe ich zusammen mit folgendem Prompt in Claude eingefügt:
Analyze the following SnpEff genetic annotations for Felis catus 9.0. When referencing a gene, make sure to mention how many mutations were discovered in that gene and do not hallucinate mutations for genes that are not provided in the annotation file pasted below. Please provide:
1. A summary of the most significant findings
2. Highlight any mutations in genes associated with known health conditions, phenotypic traits, or important biological processes.
3. Explain the potential functional implications of the top 5–10 most interesting mutations, considering the gene's function and the type of mutation.
4. Identify any patterns, clusters of mutations, or impacted biological/functional pathways that might be noteworthy.
5. Suggest any potential health concerns or traits that might be affected based on these mutations, but clarify that this is speculative without clinical correlation.
Please use scientific terminology where appropriate, but also provide layman explanations to ensure understanding. If you need any clarification or additional information about the subject's phenotype or health history to improve your analysis, please ask.
The cat has an all white coat, is 11 years old, and has no known health issues other than feline tooth resorption. The cat has no known neurological or immune system disabilities (aside from the possible link between immune system function and feline tooth resorption).
Das ergab die folgenden Resultate, die ich ziemlich spannend fand: Claude legt eine mögliche genetische Veranlagung für feline Zahnresorption nahe – basierend auf einigen Genen mit High-Impact-Mutationen. Bei einem komplett weißen Fell ist auch der Hinweis nützlich, dass möglicherweise ein erhöhtes Krebsrisiko besteht, weil die Funktion eines DNA-Reparaturproteins beeinträchtigt sein könnte. Für mich heißt das konkret, dass ich seine direkte Sonneneinstrahlung begrenzen sollte.
Claudes weitere Schlussfolgerungen sind allerdings recht spekulativ – ich habe keines der genannten ernsthaften Probleme mit Muskelfunktion oder Stoffwechsel beobachtet. Da es keine gut organisierten, öffentlich zugänglichen Annotationsdatensätze gibt, wird die Funktion von Katzengenen weitgehend über die Homologie zu ihren menschlichen Pendants abgeleitet. Daher ist zu erwarten, dass LLMs mit auf den Menschen ausgerichteten, allgemein gehaltenen Funktionsableitungen antworten – was zu den überspekulativen Ergebnissen von Claude führt.
Thank you for providing this detailed genetic information about your 11-year-old white cat with feline tooth resorption. I'll analyze the high-impact mutations based on the SnpEff annotations you've shared. Let's break this down into the requested sections:
1. Summary of Most Significant Findings:
The genetic data reveals a substantial number of high-impact mutations across various genes. These mutations include stop-gained, start-lost, and stop-lost variants, which can significantly alter protein function. In total, there are over 200 high-impact mutations identified across numerous genes.
2. Mutations in Genes Associated with Known Conditions or Traits:
a) LRRC45 (1 mutation): Associated with ciliary function, which can affect various physiological processes.
b) PNKP (1 mutation): Involved in DNA repair, potentially impacting cancer susceptibility and neurological function.
c) ATP2A1 (1 mutation): Critical for muscle function, particularly in fast-twitch muscle fibers.
d) COQ7 (1 mutation): Important for coenzyme Q10 biosynthesis, affecting energy production in cells.
e) ABCC6 (1 mutation): Associated with connective tissue disorders in humans.
f) CRP (1 mutation): Involved in immune response and inflammation.
3. Potential Functional Implications of Top Mutations:
a) PNKP (c.1150C>T, p.Gln384*): This stop-gained mutation could lead to a truncated protein, potentially impairing DNA repair mechanisms. This might increase susceptibility to DNA damage and theoretically could affect neurological function or cancer risk.
b) ATP2A1 (c.3149A>G, p.Ter1050Trpext*?): This stop-lost mutation might result in an extended protein. ATP2A1 is crucial for muscle contraction, and alterations could potentially affect muscle function.
c) COQ7 (c.817T>A, p.Ter273Lysext*?): Another stop-lost mutation, this could lead to an extended protein. COQ7 is vital for coenzyme Q10 production, which is essential for cellular energy production. Alterations might affect overall energy metabolism.
d) ABCC6 (c.4246C>T, p.Arg1416*): This stop-gained mutation could result in a truncated protein. In humans, ABCC6 mutations are associated with pseudoxanthoma elasticum, a condition affecting connective tissue.
e) CRP (c.592T>C, p.Ter198Glnext*?): This stop-lost mutation in the C-reactive protein gene could affect inflammatory responses and immune function.
4. Patterns and Noteworthy Observations:
There appears to be a cluster of mutations affecting genes involved in muscle function, energy metabolism, and immune response. Several mutations are found in genes related to DNA repair and cellular maintenance processes.
5. Potential Health Concerns or Traits (Speculative):
Based on these mutations, there might be potential concerns related to:
\* Muscle function and metabolism (ATP2A1, COQ7)
\* Immune system regulation (CRP)
\* DNA repair and potential cancer susceptibility (PNKP)
\* Connective tissue health (ABCC6)
However, it's crucial to emphasize that the presence of these mutations doesn't necessarily mean the cat will develop related conditions. Many factors, including environmental influences and other genetic modifiers, play a role in gene expression and disease development.
It's interesting to note that despite these mutations, your cat has lived to 11 years with no major health issues reported aside from feline tooth resorption. This underscores the complex relationship between genotype and phenotype.
Given the cat's history of feline tooth resorption, the mutations in genes related to immune function (like CRP) and connective tissue (like ABCC6) might be of particular interest, as these systems are thought to play a role in this condition. However, a direct link cannot be established without further research.
Im nächsten Schritt wollte ich herausfinden, welche Gene bekanntermaßen mit weißer Fellfarbe bei Katzen in Verbindung stehen, und dann nach allen von SnpEff annotierten Mutationen innerhalb dieser Gene filtern – unabhängig davon, ob SnpEff die Auswirkung als low, moderate oder high einstuft. Dazu habe ich die Annotationsdatei nach Mutationen in den Genen KIT und KITLG gefiltert:
head -n1 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff.ann.tsv > 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_LOW_MODERATE_impact_white_coat.ann.tsv
grep -E "\t(LOW|MODERATE)\t.*protein_coding" 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff.ann.tsv | grep -E '\tKIT\t|\tKITLG\t' >> 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_LOW_MODERATE_impact_white_coat.ann.tsv
cut -f 1,2,4,5,8-11,17,18,19,21 31220812113030_GCF_000181335.3_Felis_catus_9.0_genomic_snpeff_LOW_MODERATE_impact_white_coat.ann.tsv
Mit demselben Prompt vermutet Claude, dass die Missense- und Intron-Splice-Site-Mutationen wahrscheinlich für Clouds weiße Fellfarbe verantwortlich sind:
Thank you for providing the genetic annotations for Felis catus 9.0. I'll analyze this data and provide the requested information. Let's break down the findings:
1. Summary of the most significant findings:
The annotations reveal multiple mutations in two important genes: KIT and KITLG. These genes are part of the KIT/KITLG signaling pathway, which plays crucial roles in various biological processes, including coat color development, hematopoiesis, and gametogenesis.
KIT gene: 9 mutations (7 synonymous variants, 2 splice region variants)
KITLG gene: 2 mutations (1 missense variant, 1 synonymous variant)
The most significant finding is the missense variant in the KITLG gene, which could potentially affect protein function.
2. Mutations associated with known health conditions, phenotypic traits, or important biological processes:
The KIT and KITLG genes are associated with coat color in cats, particularly white spotting patterns. The all-white coat of this cat might be related to these mutations, especially considering the number of variants in the KIT gene.
3. Potential functional implications of the top 5–10 most interesting mutations:
a) KITLG missense variant (c.731G>A, p.Ser244Asn): This mutation changes a serine to an asparagine at position 244 in the KITLG protein. This could potentially affect the protein's function, as it alters the amino acid sequence. KITLG is crucial for the survival and proliferation of melanocytes, which produce pigment. This mutation might contribute to the cat's all-white coat.
b) KIT splice region variants (c.2491–3C>T, c.1353–5T>C, c.1122–4_1122–3delGT): These mutations occur in splice regions, which are important for proper mRNA processing. Although they're classified as low impact, they could potentially affect the splicing of the KIT gene, leading to altered protein production or function. The KIT protein is a receptor tyrosine kinase involved in various developmental processes, including melanocyte development.
c) KIT synonymous variants: While synonymous variants don't change the amino acid sequence, they can still impact gene expression through various mechanisms such as altering mRNA stability or translation efficiency. The high number of synonymous variants in the KIT gene (7 in total) might collectively influence its expression or function.
4. Patterns, clusters of mutations, or impacted biological pathways:
The mutations are clustered in the KIT/KITLG signaling pathway, which is crucial for melanocyte development and function. This pathway is also involved in hematopoiesis (blood cell formation) and gametogenesis (production of reproductive cells). The concentration of mutations in these genes suggests a potential impact on pigmentation and possibly other related processes.
5. Potential health concerns or traits that might be affected (speculative):
a) Coat color: The all-white coat of the cat is likely related to these mutations, particularly in the KIT and KITLG genes.
b) Hearing: Some all-white cats with KIT mutations are prone to congenital deafness. While no specific deafness-associated mutations are present here, it might be worth monitoring the cat's hearing.
c) Immune function: The KIT/KITLG pathway plays a role in hematopoiesis, including the development of certain immune cells. While the cat has no known immune system disabilities, these mutations could potentially influence immune function subtly.
d) Tooth resorption: Although not directly linked to KIT/KITLG in current literature, the pathway's involvement in various developmental processes might indirectly contribute to dental issues. This is highly speculative and would require further research to establish any potential connection.
e) Fertility: Given the role of KIT/KITLG in gametogenesis, there could be potential effects on fertility. However, this is speculative and would only be relevant if the cat were to be bred.
It's important to note that while these genetic findings are interesting, their clinical significance can only be determined through correlation with the cat's actual health status and further studies. The cat's current good health at 11 years old suggests that many of these mutations may not have severe impacts.
Mein Fazit aus all dem: Aus den Genomdaten Ihrer Katze lassen sich tatsächlich einige ziemlich coole Erkenntnisse gewinnen – und der Weg zu diesen Daten wird durch Cloud-Services, die Daten-Pipelines orchestrieren (etwa Cloud Composer), spürbar praktikabler, zuverlässiger und skalierbarer.
Auf der Seite der öffentlich finanzierten Forschung bleibt allerdings noch viel zu tun. Die Funktion von Katzengenen über ihre Homologie zu menschlichen Genen abzuleiten, ermöglicht aussagekräftige Analysen nur in begrenztem Maße. Sobald veterinärmedizinische Forschungseinrichtungen anfangen, genomische Annotationsdatenbanken für Katzen und Hunde zu veröffentlichen, werden LLM-gestützte Analysen genomischer Daten meiner Meinung nach ihr volles Potenzial entfalten. Bis dahin bleibt mir der Respekt vor der wissenschaftlichen Arbeit, die das Basepaws-Team geleistet hat.
Und damit sind wir am Ende! Wir haben einen Streifzug durch die faszinierende Welt der Katzen-Genomik unternommen – vom beeindruckenden BasePaws-Report bis zur eigenen DIY-Analyse, ermöglicht durch hochmoderne Cloud-Computing-Services und generative KI. Auch wenn das Orchestrieren einer Symphonie genomischer Tools manchmal so chaotisch sein kann wie das Hüten einer Schar Katzen, machen Services wie Cloud Composer die Sache deutlich einfacher.
PerfectScale™ for Kubernetes
Ready to optimize?
Get your free Kubernetes savings analysis
Your cloud bill shouldn't be a mystery
Optimization, automation, expertise. In one platform.